UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
For the fiscal year ended December 31, 2018
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File No. 0-21392
Amarin Corporation plc
(Exact name of registrant as specified in its charter)
|
|
|
|
England and Wales |
Not applicable |
|
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
2 Pembroke House
Upper Pembroke Street 28-32, Dublin 2, Ireland
(Address of principal executive offices)
+353 (0) 1 6699 020
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
|
Title of Each Class
|
Name of Each Exchange on Which Registered
|
|
American Depositary Shares, each representing one Ordinary Share Ordinary Shares, 50 pence par value per share |
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ☑ NO ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ☐ NO ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES ☑ NO ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). YES ☑ NO ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☑
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
|
|
|
|
Large accelerated filer |
☑ |
Accelerated filer ☐ |
|
|
|
|
|
Non-accelerated filer |
☐ |
Smaller reporting company ☐ |
|
|
|
|
|
Emerging growth company |
☐ |
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). YES ☐ NO ☑
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant as of June 30, 2018 was approximately $902.0 million, based upon the closing price on the NASDAQ Capital Market reported for such date.
329,321,488 shares were outstanding as of February 22, 2019, including 329,087,415 shares held as American Depositary Shares (ADSs), each representing one Ordinary Share, 50 pence par value per share and 234,073 Ordinary Shares. In addition, 28,931,746 ordinary share equivalents were issuable in exchange for outstanding preferred shares as of February 22, 2019, for a total of 358,253,234 ordinary shares and ordinary share equivalents outstanding as of February 22, 2019.
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required to be disclosed in Part III of this report is incorporated by reference from the registrant’s definitive proxy statement to be filed not later than 120 days after the end of the fiscal year covered by this report.
|
|
|
|
|
Page |
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 1. |
|
|
1 |
|
|
Item 1A. |
|
|
27 |
|
|
Item 1B. |
|
|
63 |
|
|
Item 2. |
|
|
63 |
|
|
Item 3. |
|
|
64 |
|
|
Item 4. |
|
|
65 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 5. |
|
|
66 |
|
|
Item 6. |
|
|
70 |
|
|
Item 7. |
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
71 |
|
Item 7A. |
|
|
87 |
|
|
Item 8. |
|
|
88 |
|
|
Item 9. |
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
|
88 |
|
Item 9A. |
|
|
88 |
|
|
Item 9B. |
|
|
91 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 10. |
|
|
|
|
|
Item 11. |
|
|
92 |
|
|
Item 12. |
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
|
92 |
|
Item 13. |
|
Certain Relationships and Related Transactions, and Director Independence |
|
92 |
|
Item 14. |
|
|
92 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 15. |
|
|
93 |
|
|
Item 16. |
|
|
99 |
|
|
|
|
|
|
|
|
|
100 |
|||
SPECIAL NOTE REGARDING
FORWARD-LOOKING STATEMENTS AND INDUSTRY DATA
This Annual Report on Form 10-K contains forward-looking statements. All statements other than statements of historical fact contained in this Annual Report on Form 10-K are forward-looking statements, including statements regarding the progress and timing of our clinical programs, regulatory filings and commercialization activities, and the potential clinical benefits, safety and market potential of our product candidates, as well as more general statements regarding our expectations for future financial and operational performance, regulatory environment, and market trends. In some cases, you can identify forward-looking statements by terminology such as “may,” “would,” “should,” “could,” “expects,” “aims,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “projects,” “potential,” or “continue”; the negative of these terms; or other comparable terminology. These statements include but are not limited to statements regarding the commercial success of Vascepa and factors that can affect such success; interpretation of court decisions; expectation on determinations and policy positions of the United States Food and Drug Administration, or FDA; the expected timing of enrollment, interim results and final results of our REDUCE-IT study; the safety and efficacy of our product and product candidates; expectation regarding the potential for Vascepa to be partnered, developed and commercialized outside of the United States; expectation on the scope and strength of our intellectual property protection and the likelihood of securing additional patent protection; estimates of the potential markets for our product candidates; estimates of the capacity of manufacturing and other facilities to support our products; our operating and growth strategies; our industry; our projected cash needs, liquidity and capital resources; and our expected future revenues, operations and expenditures.
Forward-looking statements are only current predictions and are subject to known and unknown risks, uncertainties, and other factors that may cause our or our industry’s actual results, levels of activity, performance, or achievements to be materially different from those anticipated by such statements. These factors include, among other things, those listed under “Risk Factors” in Item 1A of Part I of this Annual Report on Form 10-K and elsewhere in this Annual Report on Form 10-K. These and other factors could cause results to differ materially from those expressed in these forward-looking statements.
Although we believe that the expectations reflected in the forward-looking statements contained in this Annual Report on Form 10-K are reasonable, we cannot guarantee future results, performance, or achievements. Except as required by law, we are under no duty to update or revise any of such forward-looking statements, whether as a result of new information, future events or otherwise, after the date of this Annual Report on Form 10-K.
Unless otherwise indicated, information contained in this Annual Report on Form 10-K concerning our product candidates, the number of patients that may benefit from these product candidates and the potential commercial opportunity for our product candidates, is based on information from independent industry analysts and third-party sources (including industry publications, surveys, and forecasts), our internal research, and management estimates. Management estimates are derived from publicly available information released by independent industry analysts and third-party sources, as well as data from our internal research, and based on assumptions made by us based on such data and our knowledge of such industry, which we believe to be reasonable. None of the sources cited in this Annual Report on Form 10-K has consented to the inclusion of any data from its reports, nor have we sought their consent. Our internal research has not been verified by any independent source, and we have not independently verified any third-party information. While we believe that such information included in this Annual Report on Form 10-K is generally reliable, such information is inherently imprecise. In addition, projections, assumptions, and estimates of our future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in “Risk Factors” in Item 1A of Part I of this Annual Report on Form 10-K and elsewhere in this Annual Report on Form 10-K. These and other factors could cause results to differ materially from those expressed in the estimates made by the independent parties and by us.
References in this report to “Amarin,” the “Company,” “we,” “our” and “us” refer to Amarin Corporation plc and its subsidiaries, on a consolidated basis, unless otherwise indicated.
This Annual Report on Form 10-K includes the registered and unregistered trademarks and service marks of other parties.
Amarin Corporation plc is a public limited company incorporated under the laws of England and Wales. Amarin Corporation plc was originally incorporated in England as a private limited company on March 1, 1989 under the Companies Act 1985, and re-registered in England as a public limited company on March 19, 1993.
Our principal offices are located at 2 Pembroke House, Upper Pembroke Street 28-32, Dublin 2 Ireland. Our registered office is located at One New Change, London EC4M 9AF, England. Our primary office in the United States is located at 1430 Route 206, Bedminster, NJ 07921, USA. Our telephone number at that location is (908) 719-1315.
1
For purposes of this Annual Report on Form 10-K, our ordinary shares may also be referred to as “common shares” or “common stock.”
We are a pharmaceutical company with expertise in omega-3 fatty acids and lipid science focused on the commercialization and development of therapeutics to improve cardiovascular, or CV, health.
Our lead product, Vascepa® (icosapent ethyl) capsules, is approved by the U.S. Food and Drug Administration, or FDA, for use as an adjunct to diet to reduce triglyceride, or TG, levels in adult patients with severe (TG ≥500 mg/dL) hypertriglyceridemia. Triglycerides are the main constituent of body fat in humans. Hypertriglyceridemia refers to a condition in which patients have high levels of triglycerides in the bloodstream. The primary targeted clinical benefit of lowering triglycerides in adult patients with severe (TG ≥500 mg/dL) hypertriglyceridemia is to reduce the risk of pancreatitis. In January 2013, we began selling and marketing Vascepa in the United States based on the FDA-approved MARINE indication of patients with severely high (TG ≥500 mg/dL) triglyceride levels, a patient population of approximately 4 million people in the United States.
Our FDA-approved indication for Vascepa, known as the MARINE indication, is based primarily on the successful results from the MARINE study of Vascepa in the approved patient population. In considering this approval, the FDA also reviewed the successful results from our study of Vascepa in patients with high triglyceride levels (TG ≥200 mg/dL and <500 mg/dL) who are also on statin therapy for elevated low-density lipoprotein cholesterol, or LDL-C, levels which condition we refer to as mixed dyslipidemia or persistent high triglycerides. This study is known as the ANCHOR study.
In August 2015, in addition to our FDA-approved indication, we began promoting Vascepa to healthcare professionals, or HCPs, in the United States for the lowering of triglyceride levels and other lipid and lipoprotein parameters in treatment of the patient population studied in the ANCHOR study (persistent high triglycerides after statin therapy). It is estimated that one in four adults in the United States, or more than 50 million people, have elevated (>150 mg/dL) triglyceride levels. We also educated HCPs with supportive but not conclusive early stage and Japanese cardiovascular outcomes trial research on how the unique active ingredient in Vascepa, icosapent ethyl, might reduce the risk of coronary heart disease. This HCP promotion was based on an August 2015 federal court declaration and subsequent settlement with the FDA and U.S. government that we believe permits such promotion under the freedom of speech clause of the First Amendment to the United States Constitution. To remain truthful and non-misleading, as part of this promotion we educated HCPs on the continued uncertainty between lowering triglycerides and cardiovascular risk reduction based on the failure of other drugs (fenofibrate and formulations of niacin) to demonstrate incremental cardiovascular benefit from adding a second lipid-altering drug on top of standard of care statin therapy, despite such drugs reducing triglyceride levels and having other favorable effects on lipid and lipoprotein parameters. We believe that, in general, HCPs prefer to review and rely on robust cardiovascular outcomes trial results before changing prescribing practices.
Multiple primary and secondary prevention trials have shown a significant relative risk reduction, or RRR, of 25% to 35% in the risk of cardiovascular events with statin therapy, leaving significant persistent residual risk despite the achievement of target LDL-C levels. Worldwide, cardiovascular disease, or CVD, remains the number one killer of men and women. In the United States, CVD leads to one in every three deaths—one death approximately every 38 seconds—with annual treatment cost in excess of $500 billion. There is no FDA-approved therapy for lowering cardiovascular risk beyond therapies which target lowering of LDL-C levels.
Since our inception, we have devoted substantial resources to our research and development efforts, most significantly our Vascepa cardiovascular outcomes trial, REDUCE-IT™, which we commenced in 2011 and was conducted based on a special protocol assessment, or SPA, agreement with the FDA. REDUCE-IT was a global study of 8,179 statin-treated adults with elevated cardiovascular risk. We announced topline results from the REDUCE-IT study on September 24, 2018. On November 10, 2018, we announced the more detailed, primary results from the REDUCE-IT study at the 2018 Scientific Sessions of the American Heart Association, or AHA, and the results were concurrently published in The New England Journal of Medicine. REDUCE-IT met its primary endpoint demonstrating a 25% relative risk reduction, or RRR, to a high degree of statistical significance (p<0.001), in first occurrence of major adverse cardiovascular events, or MACE, in the intent-to-treat patient population with use of Vascepa 4 grams/day as compared to placebo. Patients who were enrolled in REDUCE-IT needed to have LDL-C between 41-100 mg/dL (median baseline LDL-C75 mg/dL) controlled by statin therapy and various cardiovascular risk factors including persistent elevated triglycerides, or TG, between 135-499 mg/dL (median baseline 216 mg/dL) and either established cardiovascular disease (secondary prevention cohort) or be at least age 50 with diabetes mellitus and at least one other CV risk factor (primary prevention cohort). Approximately 59% of the patients had diabetes at baseline, approximately 71% of the patients had established cardiovascular disease at time of enrollment and approximately 29% were primary prevention subjects at high risk for cardiovascular disease. REDUCE-IT also showed a 26% RRR in its key secondary composite endpoint of cardiovascular death, heart attacks and stroke (p<0.001).
In September 2018, in connection with the public release of topline REDUCE-IT results, we commenced communications to healthcare professionals which were intended to ensure we meet our continuing obligation to update HCPs regarding off-label use of
2
Vascepa to ensure that our communications remain truthful and non-misleading. We believe this promotion is consistent with the federal court approved settlement with the FDA and U.S. government. After publication of primary results of the REDUCE-IT study in The New England Journal of Medicine and the scientific presentation of REDUCE-IT results at the 2018 Scientific Sessions of the American Heart Association (AHA) on November 10, 2018, we updated and expanded our communication of REDUCE-IT results to include the publication and the peer-reviewed information presented in an effort to further ensure that our communications remain truthful and non-misleading. While we believe we are now permitted under the settlement and our First Amendment rights to more broadly promote Vascepa, the FDA-approved labeling for Vascepa has not changed as we have not yet applied for FDA approval for marketing claims related to REDUCE-IT. Also, neither government nor other third-party coverage or reimbursement to pay for the off-label use of Vascepa promoted was covered under the court declaration or settlement. We plan to submit a supplemental new drug application, or sNDA, to the FDA seeking revised labeling for Vascepa based on results of the REDUCE-IT study and, upon such expanded labeling, subject to FDA approval of such label, to further expand its promotion of Vascepa in the United States.
We sell Vascepa principally to a limited number of major wholesalers, as well as selected regional wholesalers and specialty pharmacy providers, or collectively, our Distributors or our customers, that in turn resell Vascepa to retail pharmacies for subsequent resale to patients and healthcare providers. We market Vascepa in the United States through our direct sales force. In March 2014, we entered into a co-promotion agreement in the United States with Kowa Pharmaceuticals America, Inc. under which Kowa Pharmaceuticals America, Inc. began to co-promote Vascepa in conjunction with its promotion of its primary product, a branded statin for patients with high cholesterol, which commenced in May 2014 and extended until the end of 2018. Amarin and Kowa Pharmaceuticals America, Inc. intentionally designed the co-promotion to naturally end as of December 31, 2018 and mutually agreed not to renew the agreement. Prior to the REDUCE-IT results topline announcement in September 2018, our direct sales force consisted of approximately 170 sales professionals, including sales representatives and their managers. We have recently increased the size of our sales force to approximately 440 sales professionals, including approximately 400 sales representatives, in the United States pursuant to positive REDUCE-IT results and are expanding our promotion of Vascepa. Such promotion, prior to results of the REDUCE-IT study, was based on demonstrated changes in biomarkers based on our MARINE and ANCHOR studies. Most healthcare professionals express that they prefer outcomes data to biomarker data. Because prior to results of the REDUCE-IT study we did not have outcomes data regarding the clinical effect of Vascepa and because a substantial portion of our resources were being spent on the REDUCE-IT study, prior to REDUCE-IT results our commercialization of Vascepa was somewhat limited. Subsequent to learning the positive cardiovascular outcomes results of the REDUCE-IT study, we have begun increasing our promotion of Vascepa.
In addition to promotion of Vascepa in the United States, we have entered into strategic partnerships and license arrangements in Asia, the Middle East, North Africa and Canada to further promote, develop and commercialize Vascepa. In February 2015, we entered into an exclusive agreement with Eddingpharm (Asia) Macao Commercial Offshore Limited, or Eddingpharm, to develop and commercialize Vascepa capsules in Mainland China, Hong Kong, Macau and Taiwan, or the China Territory. In March 2016, we entered into an agreement with Biologix FZCo, or Biologix, to register and commercialize Vascepa in countries within the Middle East and North Africa. In September 2017, we entered into an agreement with HLS Therapeutics Inc., or HLS, to register, commercialize and distribute Vascepa in Canada.
In June 2018, we entered into a collaboration with Mochida Pharmaceutical Co., Ltd., or Mochida, related to development and potential subsequent commercialization of drug products and indications based on the active pharmaceutical ingredient in Vascepa, the omega-3 acid, EPA (eicosapentaenoic acid). The potential new product and indication opportunities contemplated under this agreement are currently in early stages of development.
We continue to assess other collaboration opportunities to maximize the value of the Vascepa franchise globally.
Commercialization—United States
We commenced the commercial launch of 1-gram size Vascepa capsules in the United States in January 2013. We commenced sales and shipments of Vascepa at that time to our network of U.S.-based wholesalers. Prior to the REDUCE-IT results topline announcement in September 2018, our direct sales force consisted of approximately 170 sales professionals, including sales representatives and their managers. We have recently increased the size of our sales force to approximately 440 sales professionals, including approximately 400 sales representatives, pursuant to positive REDUCE-IT results and are expanding our promotion of Vascepa. Commencing in May 2014, in addition to Vascepa promotion by our sales representatives, Kowa Pharmaceuticals America, Inc. began co-promoting Vascepa in conjunction with its promotion of its primary product, a branded statin for patients with high cholesterol. Amarin and Kowa Pharmaceuticals America, Inc. intentionally designed the co-promotion to naturally end as of December 31, 2018 and mutually agreed not to renew the agreement. We also employ various medical affairs and marketing personnel to support our commercialization of Vascepa. We expanded certain medical education and market awareness initiatives following the reporting of positive REDUCE-IT results in 2018. We intend to further expand promotion of Vascepa following label expansion of Vascepa, subject to FDA approval of such expanded label.
3
In October 2016, in addition to the original 1-gram capsule size for Vascepa, we introduced a smaller 0.5-gram capsule size, the first and only 0.5-gram prescription omega-3 alternative available on the market, for the subset of patients who prefer a smaller capsule. The FDA-approved dosing for Vascepa continues to be 4 grams per day, and, as expected, the majority of new and existing patients taking Vascepa continue to be prescribed the 1-gram size Vascepa capsule.
From May 2014 until December 2018, under our co-promotion agreement with Kowa Pharmaceuticals America, Inc., both parties agreed to use commercially reasonable efforts to promote, detail and optimize sales of Vascepa in the United States and agreed to specific performance requirements detailed in the related agreement. The performance requirements included a negotiated minimum number of sales details to be delivered by each party in the first and second position, the use of a negotiated number of minimum sales representatives from each party, and the achievement of minimum levels of Vascepa revenue in 2015 and beyond. First position referred to when a sales representative’s primary purpose in detailing is related to Vascepa, while second position referred to when a sales representative’s primary purpose in detailing is to promote another product, but they also devoted time in the same sales call to promote Vascepa. Kowa Pharmaceuticals America, Inc. also agreed to bear the costs incurred for its sales force associated with the commercialization of Vascepa and to pay for certain incremental costs associated with the use of its sales force, such as sample costs and costs for promotional and marketing materials. We recognized all revenue from sales of Vascepa. In exchange for Kowa Pharmaceuticals America, Inc.’s co-promotional services, Kowa Pharmaceuticals America, Inc. was entitled to a quarterly co-promotion fee based on a percentage of aggregate Vascepa gross margin that varied during the term. The percentage of aggregate Vascepa gross margin earned by Kowa Pharmaceuticals America, Inc. was, as amended, approximately eighteen percent (18%) in 2017, partially offset by certain other refinements. During 2018, which was the last year of the agreement, as amended, we incurred expense for both the annual co-promotion fee, which in 2018 was calculated as eighteen-and-a-half percent (18.5%) of Vascepa gross margin, plus accrual for co-promotion tail payments which are calculated as a percentage of the 2018 co-promotion fee. Kowa Pharmaceuticals America, Inc. is eligible to receive $17.8 million in co-promotion tail payments, the present value of which of $16.6 million was fully accrued as of December 31, 2018. The accrued tail payments will be paid over three years with declining amounts each year beginning with $7.3 million to be paid in 2019.
Based on monthly compilations of data provided by a third party, Symphony Health, the estimated number of normalized total Vascepa prescriptions for the three months ended December 31, 2018 was approximately 539,000 compared to 458,000, 430,000, 391,000, and 404,000 in the three months ended September 30, 2018, June 30, 2018, March 31, 2018, and December 31, 2017, respectively. According to data from another third party, IQVIA, the estimated number of normalized total Vascepa prescriptions for the three months ended December 31, 2018 was approximately 538,000 compared to 457,000, 430,000, 392,000, and 409,000 in the three months ended September 30, 2018, June 30, 2018, March 31, 2018, and December 31, 2017, respectively. Normalized total prescriptions represent the estimated total number of Vascepa prescriptions dispensed to patients, calculated on a normalized basis (i.e., one month’s supply, or total capsules dispensed multiplied by the number of grams per capsule divided by 120 grams). Inventory levels at wholesalers tend to fluctuate based on seasonal factors, prescription trends and other factors.
The data reported above is based on information made available to us from third-party resources and may be subject to adjustment and may overstate or understate actual prescriptions. Timing of shipments to wholesalers, as used for revenue recognition purposes, and timing of prescriptions as estimated by these third parties may differ from period to period. Although we believe this data is prepared on a period-to-period basis in a manner that is generally consistent and that such results can be generally indicative of current prescription trends, the data is based on estimates and should not be relied upon as definitive. While we expect to be able to grow Vascepa revenues over time, no guidance should be inferred from the operating metrics described above. We also anticipate that such sales growth will be inconsistent from period to period. We believe that investors should view the above-referenced operating metrics with caution, as data for this limited period may not be representative of a trend consistent with the results presented or otherwise predictive of future results. Seasonal fluctuations in pharmaceutical sales, for example, may affect future prescription trends of Vascepa, as could changes in prescriber sentiment, quarterly changes in Distributor purchases, and other factors. We believe investors should consider our results over several quarters, or longer, before making an assessment about potential future performance.
The commercialization of pharmaceutical products is a complex undertaking, and our ability to effectively and profitably commercialize Vascepa will depend in part on our ability to generate market demand for Vascepa through education, marketing and sales activities, our ability to achieve market acceptance of Vascepa, our ability to generate product revenue and our ability to receive adequate levels of reimbursement from third-party payers. See “Risk Factors—Risks Related to the Commercialization and Development of Vascepa.”
In August 2015, we and our co-promotion partner began communicating promotional information beyond MARINE clinical trial data to targeted healthcare professionals. Such qualified communications are being made pursuant to the August 2015 federal district court declaration and related March 2016 settlement allowing truthful and non-misleading promotion of the FDA-reviewed and agreed effects of Vascepa demonstrated in the ANCHOR clinical trial and presentation of the current state of scientific research related to the potential of Vascepa to reduce the risk of cardiovascular disease including through use of peer-reviewed scientific publications of available data.
4
In September 2018, in connection with the public release of topline REDUCE-IT results, we commenced communications to healthcare professionals, or HCPs, which were intended to ensure we meet our continuing obligation to update HCPs regarding off-label use of Vascepa to ensure that our communications remain truthful and non-misleading. We believe this promotion is consistent with the federal court approved settlement with the FDA and U.S. government. After publication of primary results of the REDUCE-IT study in The New England Journal of Medicine and the scientific presentation of REDUCE-IT results at the 2018 Scientific Sessions of the American Heart Association (AHA) on November 10, 2018, we updated and expanded our communication of REDUCE-IT results to include the publication and the peer-reviewed information presented in an effort to further ensure that our communications remain truthful and non-misleading. While we believe we are now permitted under the settlement and our First Amendment rights to more broadly promote Vascepa, the FDA-approved labeling for Vascepa has not changed as we have not yet applied for FDA approval for marketing claims related to REDUCE-IT. Also, neither government nor other third-party coverage or reimbursement to pay for the off-label use of Vascepa promoted was covered under the court declaration or settlement.
Commercialization—Outside the United States
In February 2015, we announced an exclusive agreement with Eddingpharm to develop and commercialize Vascepa capsules in what we refer to as the China Territory, consisting of the territories of Mainland China, Hong Kong, Macau and Taiwan, for uses that are currently commercialized and under development by us in the United States based on the MARINE, ANCHOR and REDUCE-IT clinical trials of Vascepa.
Under the agreement, Eddingpharm is responsible for development and commercialization activities in the China Territory and associated expenses. We will provide development assistance and be responsible for supplying the product. Terms of the agreement include up-front and milestone payments to us of up to $169.0 million, including a non-refundable $15.0 million up-front payment received at closing and a non-refundable milestone payment of $1.0 million received upon successful submission of a clinical trial application, or CTA, with respect to the MARINE indication for Vascepa to the Chinese regulatory authority in March 2016. In March 2017, the CTA was approved by the Chinese regulatory authority and, in December 2017, Eddingpharm commenced a pivotal clinical trial aimed to support the regulatory approval of the first indication of Vascepa in a patient population with severe hypertriglyceridemia in Mainland China. We are also entitled to receive future regulatory and sales-based milestone payments of up to an additional $153.0 million. The regulatory milestone events relate to the submission and approval of certain applications to the applicable regulatory authority, such as a clinical trial application, clinical trial exemption, or import drug license application. The amounts to be received upon achievement of the regulatory milestone events relate to the submission and approval for three indications, and range from $2.0 million to $15.0 million for a total of $33.0 million. The sales-based milestone events occur when annual aggregate net sales of Vascepa in the territory equals or exceeds certain specified thresholds, and range from $5.0 million to $50.0 million for a total of $120.0 million. Eddingpharm will also pay us tiered double-digit percentage royalties on net sales of Vascepa in the China Territory escalating to the high teens. We will supply finished product to Eddingpharm under negotiated terms.
In March 2016, we entered into an agreement with Biologix FZCo, or Biologix, to register and commercialize Vascepa in several Middle Eastern and North African countries. Under the terms of the distribution agreement, we granted to Biologix a non-exclusive license to use our trademarks in connection with the importation, distribution, promotion, marketing and sale of Vascepa in the Middle East and North Africa territory. Upon closing of the agreement, we received a non-refundable up-front payment, which will be recognized as revenue over 10 years commencing upon first marketing approval of Vascepa in the territory. We receive all payments based on total product sales and pay Biologix a service fee in exchange for its services, whereby the service fee represents a percentage of gross selling price which is subject to a minimum floor price. In March 2018 and July 2018, we received approval for Vascepa as a prescription medication for use in Lebanon and United Arab Emirates, respectively, as an adjunct to diet to reduce triglyceride levels in adult patients with severe hypertriglyceridemia.
In September 2017, we entered into an agreement with HLS to register, commercialize and distribute Vascepa in Canada. Under the agreement, HLS will be responsible for regulatory and commercialization activities and associated costs. We will be responsible for providing assistance towards local filings, supplying finished product under negotiated supply terms, maintaining intellectual property, and continuing the development and funding of REDUCE-IT related activities. Terms of the agreement include up-front and milestone payments to us of up to $65.0 million. These payments include a non-refundable $5.0 million up-front payment received in two equal installments, the first of which was received at closing with the second received upon the six-month anniversary of the closing, as well as a non-refundable milestone payment of $2.5 million received upon achievement of the REDUCE-IT trial primary endpoint. In addition to the non-refundable, up-front payment, we are entitled to receive certain regulatory and sales-based milestone payments of up to an additional $57.5 million, the timing and achievability of which cannot be determined at this time, as well as tiered double-digit royalties on net sales of Vascepa in Canada.
We continue to assess other partnership opportunities for licensing Vascepa to partners outside of the United States.
5
Since our inception, we have devoted substantial resources to the research and development of Vascepa (icosapent ethyl) capsules. Vascepa is a single-molecule prescription product consisting of the omega-3 acid commonly known as EPA in ethyl-ester form. Vascepa is derived from fish through a stringent and complex FDA-regulated manufacturing process designed to effectively eliminate impurities and isolate and protect the single molecule active ingredient from degradation. Vascepa has been designated a new chemical entity by the FDA. Vascepa is known in scientific literature as AMR101.
Our most important clinical trials of Vascepa are summarized here and discussed in further detail below:
|
|
• |
The ANCHOR trial, a Phase 3 multi-center, placebo-controlled, randomized, double-blind, 12-week pivotal study in patients with high (≥200 and <500 mg/dL) TGs who were also receiving optimized statin therapy with the primary endpoint being the lowering of TG levels; and |
|
|
• |
The REDUCE-IT trial, a Phase 3 global study of 8,179 statin-treated adults with elevated cardiovascular risk with a primary endpoint being the first occurrence of major adverse cardiovascular events, or MACE, in the intent-to-treat patient population, patients with LDL-C between 41-100 mg/dL (median baseline LDL-C75 mg/dL) controlled by statin therapy and various cardiovascular risk factors including persistent elevated TG between 135-499 mg/dL and either established cardiovascular disease (secondary prevention cohort) or age 50 or more with diabetes mellitus and at least one other CV risk factor (primary prevention cohort). |
The REDUCE-IT cardiovascular outcomes study of Vascepa has been the centerpiece of our research and development efforts. Prior research on Vascepa, such as the MARINE and ANCHOR trials, had been focused on the effects of the drug on biomarkers associated with increased risk of pancreatitis and increased risk of cardiovascular events. Other prior and ongoing research and development efforts include the study of potential mechanisms of action of Vascepa.
In June 2018, we entered into a multi-faceted collaboration with Mochida related to the development and commercialization of drug products and indications based on the active pharmaceutical ingredient in Vascepa, the omega-3 acid, EPA. Among other terms in the agreement, we obtained an exclusive license to certain Mochida intellectual property to advance our interests in the United States and certain other territories and the parties will collaborate to research and develop new products and indications based on EPA for our commercialization in the United States and certain other territories. The potential new product and indication opportunities contemplated under this agreement are currently in early stages of development.
Additional research and development opportunities beyond REDUCE-IT will be assessed more fully after giving priority to securing regulatory approval for Vascepa based on the REDUCE-IT results.
Commercial and Clinical Supply
We manage the manufacturing and supply of Vascepa internally and have done so since we began clinical development of Vascepa prior to the drug’s marketing approval by the FDA in 2012. We rely on contract manufacturers in each step of our commercial and clinical product supply chain. These steps include active pharmaceutical ingredient, or API, manufacturing, encapsulation of the API, product packaging and supply-related logistics. Our approach to product supply procurement is designed to mitigate risk of supply interruption and maintain an environment of cost competition through diversification of contract manufacturers at each stage of the supply chain and lack of reliance on any single supplier. We have multiple FDA-approved international API suppliers, encapsulators and packagers to support the Vascepa commercial franchise. The amount of supply we seek to purchase in future periods will depend on the level of growth of Vascepa revenues and minimum purchase commitments with certain suppliers. While our current supply chain is scalable, we continue efforts to expand, diversify and further enhance it.
Financial Position
We believe that our cash and cash equivalents of $249.2 million as of December 31, 2018 will be sufficient to fund our projected operations for at least twelve months and through the likely Prescription Drug User Fee Act (PDUFA) date for approval of a supplemental new drug application (sNDA) by the FDA based on REDUCE-IT study results. Depending on the level of cash generated from operations, and depending in part on the timing and results of the FDA review of the sNDA and rate of prescription growth for Vascepa, additional capital may be required to support planned expansion of Vascepa promotion and potential Vascepa promotion beyond which we are currently executing. If additional capital is required and we are unable to obtain additional capital, we may be forced to delay, limit or eliminate certain promotional activities. We anticipate that quarterly net cash outflows in future periods will be variable.
6
Lipid Disorders and Cardiovascular Disease
Heart attacks, strokes and other cardiovascular events represent the leading cause of death and disability among men and women in western societies. According to the Heart Disease and Stroke Statistics—2018 Update from the American Heart Association, more than 1 out of every 3 adults in the United States (approximately 92 million) currently lives with one or more types of cardiovascular disease; an estimated 1 million new or recurrent coronary events and 795,000 new or recurrent strokes occur each year; an estimated 29 million adults ≥20 years of age have high total serum cholesterol levels (≥240 mg/dL), and an estimated 71 million adults ≥20 years of age have borderline high or high low-density lipoprotein (“bad”) cholesterol, or LDL-C, levels (≥130 mg/dL).
In addition to cholesterol, lipoproteins such as LDL also carry fats in the form of triglycerides. Hypertriglyceridemia refers to a condition in which patients have high levels of triglycerides in the bloodstream and has been reported to be an independent risk factor for cardiovascular disease. Triglyceride levels provide important information as a marker associated with the risk for heart disease and stroke.
Guidelines for the management of very high triglyceride levels (≥500 mg/dL) suggest that reducing triglyceride levels is the primary treatment goal in these patients to reduce the risk of acute pancreatitis. Treating LDL-C remains an important secondary goal. Other important parameters to consider in patients with very high triglycerides include levels of apolipoprotein B (apo B), non-HDL-C, and very low-density lipoprotein cholesterol (VLDL-C). The effect of Vascepa on the risk for pancreatitis in patients with hypertriglyceridemia has not been determined.
It is estimated that over 25 million adults in the United States have elevated triglyceride levels ≥200 mg/dL and that more than 50 million adults in the United States have elevated triglyceride levels ≥150 mg/dL. Additionally, approximately 2 to 3 million adults in the United States have very high triglyceride levels (≥500 mg/dL). Since 1976, mean triglyceride levels have increased, in concert with the growing epidemic of obesity, insulin resistance, and type 2 diabetes mellitus. In contrast, mean LDL-C levels have decreased.
Mixed dyslipidemia refers to a condition in which patients have a combination of two or more lipid abnormalities including elevated triglycerides, low HDL-C, and/or elevated LDL-C. Both hypertriglyceridemia and mixed dyslipidemia are components of a range of lipid disorders collectively referred to as dyslipidemia. Dyslipidemia has been linked to atherosclerosis, commonly referred to as hardening of the arteries.
Limitations of Current Therapies
Hypertriglyceridema (HTG) is a prevalent lipid disorder in approximately 25% of the U.S. adult population. Both epidemiological and genetic data have shown associations between HTG and coronary heart disease. Many of those patients are taking statin therapy directed at lowering the risk of CVD by lowering their LDL-C levels, primarily. Recently, real world administrative database analyses have reported an increased CVD risk as well as direct healthcare costs associated with HTG despite statin therapy and controlled LDL-C compared to those with TG<150 mg/dL. It is estimated that approximately 6% or less of U.S. adults with triglyceride levels ≥200 mg/dL are currently receiving prescription medication for lowering triglycerides, many of whom are already on statin therapy.
Vascepa is not FDA-approved to lower TG levels in statin-treated patients with mixed dyslipidemia and persistent high (≥200 mg/dL and <500 mg/dL) TG levels due to uncertainty raised by FDA in 2013 regarding the benefit, if any, of drug-induced changes in lipid/lipoprotein parameters beyond statin-lowered LDL-C on cardiovascular risk among statin-treated patients with residually high TG. This lack of approval by FDA based on the pharmaceutical target of lowering TG levels as a surrogate for lowering the risk of CVD was due primarily to the failure of cardiovascular outcomes trials of lipid altering drugs in the fenofibrate and niacin drug classes. In CV outcomes trials, therapies that reduce TG levels and had other favorable effects on classically studied lipid and lipoprotein parameters, such as extended-release niacin and fibrates, did not met their primary CV endpoints to reduce risk when taken with contemporary medical therapy, including statins. Specifically, cardiovascular outcomes trials, ACCORD Lipid, AIM-HIGH, and HPS2-THRIVE, while not designed to test the effect of lowering TG levels in patients with high TG levels after statin therapy, each failed to demonstrate incremental cardiovascular benefit of adding a second lipid-altering drug (fenofibrate or formulations of niacin), despite raising HDL-C and reducing TG levels, among statin-treated patients with well-controlled LDL-C. As a result, in 2015, the FDA updated both the Trilipix® (a fenofibrate) and extended-release niacin product labeling and removed combination use with statin therapy in mixed dyslipidemia patients as an indication due to a failed outcomes trial. No head-to head, randomized, well-controlled studies have been conducted to compare the effects of Vascepa with other FDA-approved TG-lowering therapies.
Additionally, recent CV outcomes trials and meta-analyses with low dose omega-3 fatty acid mixtures containing DHA have not shown substantial benefit in patients receiving contemporary medical therapy, including statins. Due to these failed low dose omega-3 CV outcomes trials, the European regulatory authorities have concluded that omega-3 fatty acid medicines (specifically Lovaza®/Omacor®) at a dose of 1-gram per day are not effective in preventing further events for patients who have had a heart attack.
7
It is against that backdrop of failed studies demonstrating the limitations of other add-on drugs to statin drug therapies that we conducted the REDUCE-IT cardiovascular outcomes trial.
Potential Benefits and Market Opportunity for Vascepa
Vascepa is 1-gram of icosapent ethyl, or ethyl-EPA, and contains no DHA. We believe that the removal of DHA mitigates against the LDL-C raising effect observed in omega-3 compositions that include DHA. Based on the results of the MARINE trial, Vascepa was the first omega-3 based product to demonstrate statistically significant triglyceride reduction without a statistically significant increase in LDL-C in this very high triglyceride population.
We believe that the results of the REDUCE-IT, ANCHOR and MARINE clinical trials of Vascepa and Vascepa’s EPA only/DHA-free composition position Vascepa to achieve a global “best-in-class” prescription therapy in studied patient populations. Potential mechanisms of action at work in the reduction of cardiovascular events seen in REDUCE-IT as discussed in The New England Journal of Medicine publication of REDUCE-IT primary results include TG reduction, anti-thrombotic effects, antiplatelet or anticoagulant effects, membrane-stabilizing effects, effects on stabilization and/or regression of coronary plaque and inflammation reduction. Mechanisms responsible for the benefit shown in REDUCE-IT were not studied in REDUCE-IT as that was not the purpose of an outcomes study. More study is needed to determine to what extent, if any, each of these effects or others may be responsible for the CV risk reduction benefit demonstrated in REDUCE-IT.
The MARINE Trial (basis for currently FDA-approved label for Vascepa)
The MARINE trial, the largest study ever conducted with the omega-3 fatty acid ethyl EPA in treating patients with very high triglycerides (≥500 mg/dL), was a Phase 3, multi-center, placebo-controlled, randomized, double-blind, 12-week study. Patients were randomized into three treatment arms for treatment with Vascepa 4 gram/day, 2 gram/day or placebo. Patient enrollment in this trial began in December 2009, and enrollment and randomization was completed in August 2010 at 229 patients. The primary endpoint in the trial was the percentage change in triglyceride level from baseline compared to placebo after 12 weeks of treatment. The MARINE study primary endpoint was required to meet a stringent level of statistical significance of 1% (p < 0.01) in our special protocol assessment, or SPA, agreement with the FDA.
In November 2010, we reported topline data for the MARINE trial. In the trial, Vascepa met its primary endpoint at doses of 4 grams and 2 grams per day with median placebo-adjusted reductions in triglyceride levels of 33% (p < 0.0001) compared to placebo for 4 grams and 20% (p = 0.0051) compared to placebo for 2 grams. The median baseline triglyceride levels were 703 mg/dL, 680 mg/dL and 657 mg/dL for the patient groups treated with placebo, 4 grams of Vascepa and 2 grams of Vascepa, respectively.
In a pre-specified secondary analysis in the subgroup of patients with baseline triglyceride > 750 mg/dL, representing 39% of all patients, the effect of Vascepa in reducing triglyceride levels compared to placebo was 45% for 4 grams and 33% for 2 grams, both statistically significant (p = 0.0001 for 4 grams and p= 0.0016 for 2 grams, respectively). The median baseline triglyceride levels in this subgroup were 1052 mg/dL, 902 mg/dL and 948 mg/dL for placebo, 4-gram and 2-gram groups, respectively. Twenty-five percent of patients in this trial were also on background statin therapy. These patients had greater median reduction in triglyceride levels, which was also statistically significant.
Importantly, the significant reduction in triglycerides was not associated with a statistically significant increase in median LDL-C compared to placebo at either dose (-2.3% for the 4-gram group and +5.2% for the 2-gram group [both p=NS]). In addition, there was a statistically significant decrease in median non-HDL-C (total cholesterol less so-called “good cholesterol”) compared to placebo with both of the Vascepa-treated groups (-18% for the 4-gram group [p < 0.001] and -8% for the 2-gram group [p < 0.05]).
The MARINE trial results also included statistically significant reductions compared to placebo in several important lipid and inflammatory biomarkers, including apo B (apolipoprotein B) (8.5%), Lp-PLA2 (lipoprotein-phospholipase A2) (13.6%), VLDL-C (very low-density lipoprotein cholesterol) (28.6%), Total Cholesterol (16.3%), and hsCRP (high-sensitivity C-reactive protein) (36.0%) at the 4-gram dose. For these achieved endpoints, p-values were <0.01 for most and <0.05 for all. Apo B (apolipoprotein B) is believed to be a sensitive biomarker of cardiovascular risk and may be a better predictor of cardiovascular risk than LDL-C. Lp-PLA2 is an enzyme found in blood and atherosclerotic plaque; high levels have been implicated in the development and progression of atherosclerosis. In a post-hoc analysis of MARINE study data, Vascepa 4 g/day and 2 g/day statistically significantly reduced ApoC-III levels by 25.1% (p < 0.0001) and 14.3% (p=0.0154) versus placebo, respectively. In the MARINE trial, patients treated with 4 grams per day of Vascepa experienced a significant reduction in median placebo-adjusted lipoprotein particle concentrations of total LDL and small LDL. When looking at lipoprotein particle concentrations and sizes as measured with nuclear magnetic resonance spectroscopy, Vascepa 4 grams per day, compared with placebo, significantly reduced median total LDL particle count by 16.3% (p=0.0006), which is an important factor in atherogenesis. LDL particle count and apo B are important risk markers for the prediction of cardiovascular events. Small LDL particle count, which is a common risk factor for cardiovascular events in patients with diabetes,
8
was reduced by 25.6% (p<0.0001) compared with placebo. Vascepa 2 grams per day, compared with placebo, significantly reduced median small LDL particle count by 12.8% (p <0.05) and reduced median total LDL particle count by 1.1% (NS). LDL particle size did not change significantly for the 2 or 4 gram per day doses.
Vascepa was well tolerated in the MARINE trial, with a safety profile comparable to placebo and there were no treatment-related serious adverse events observed. No patient discontinued treatment of Vascepa during this study due to Vascepa-related adverse events. No significant changes in fasting blood glucose, hemoglobin A1C, vital signs, electrocardiograms, or liver or kidney function were observed with either Vascepa dose.
Patients enrolled in the MARINE trial were given the option to be treated with Vascepa for a period of up to 40 weeks after their last dose in the double-blind portion of the trial. Once participants completed the randomized, double blind, placebo-controlled 12-week MARINE registration trial, patients in all three randomized groups (4 grams, 2 grams and placebo) were offered the opportunity to participate in the open label extension, or OLE, phase. Patients in the OLE phase received 4 grams per day of Vascepa for a period of up to an additional 40 weeks. As is typical of such extension phases, the OLE phase was not a controlled trial, as differentiated from the randomized, double blind, placebo-controlled 12-week MARINE registration trial. In the OLE phase, participants were not randomized at entry, Vascepa administration was open-label (and thus not blinded), and no placebo group was maintained. Also, once patients entered in the OLE phase, investigators were free to add or modify other lipid-altering nutritional, lifestyle and drug treatment regimens. Given the lack of randomization, the open-label design, the addition of various other lipid-altering drugs and changes to doses of existing lipid-altering drugs, as well as the lack of placebo control, neither we nor our independent advisors were able to draw efficacy conclusions from the data. However, we have concluded that the MARINE OLE phase revealed no new safety signals after an additional 40 weeks of exposure to Vascepa, whether used alone or in combination with other lipid-altering regimens.
The ANCHOR Trial (promoted in the United States under court declaration)
The ANCHOR trial was a multi-center, placebo-controlled, randomized, double-blind, 12-week pivotal study in patients with high triglycerides (≥200 and <500 mg/dL) who were also receiving optimized statin therapy. Patients were randomized into three arms for treatment with Vascepa 4 gram/day, 2 gram/day or placebo. Patient enrollment in this trial began in January 2010, and enrollment and randomization was completed in February 2011 at 702 patients. The primary endpoint in the trial was the percentage change in triglyceride level from baseline compared to placebo after 12 weeks of treatment.
In April 2011, we reported topline results from the ANCHOR trial. The ANCHOR trial met its primary endpoint at doses of 4 grams and 2 grams per day with median placebo-adjusted reductions in triglyceride levels of 21.5% (p<0.0001 value) for 4 grams and 10.1% (p=0.0005) for 2 grams. The median baseline triglyceride levels were 259 mg/dL, 265 mg/dL and 254 mg/dL for the patient groups treated with placebo, 4 grams and 2 grams of Vascepa per day, respectively. The analysis of subgroups by baseline triglyceride tertiles showed that higher baseline triglycerides resulted in greater triglyceride reductions.
One of the trial’s secondary endpoints was to demonstrate a lack of elevation in LDL-C, the primary target of cholesterol lowering therapy. The trial’s non-inferiority criterion for LDL-C was met at both Vascepa doses. The upper confidence boundaries for both doses were below the pre-specified +6% LDL-C threshold limit. At the 4-gram dose the upper confidence boundary was below zero (-1.7%) and at the 2-gram dose the upper confidence boundary was close to zero (0.5%). For the 4 grams per day group, LDL-C decreased significantly by 6.2% from baseline versus placebo, demonstrating superiority over placebo (p=0.0067). For the 2-gram group, LDL-C decreased by 3.6% from baseline versus placebo (p=0.0867), which is not a statistically significant decrease.
Other secondary efficacy endpoints included the median placebo-adjusted percent change in non-high-density lipoprotein cholesterol (non-HDL-C), apolipoprotein B (apo B), and lipoprotein-associated phospholipase A2 (Lp-PLA2). The 4-gram dose was associated with statistically significant reductions in non-HDL-C (13.6%, p<0.0001), apo B (9.3%, p<0.0001), Lp-PLA2 (19%, p<0.0001) and high-sensitivity C-reactive protein (hsCRP) (22%, p<0.001), at week 12 compared to placebo. One published analysis showed that the Vascepa 4-gram daily dose in the ANCHOR study also significantly decreased levels of the inflammatory marker oxidized low-density lipoprotein relative to placebo by 13% (p < 0.0001). In a separate, post-hoc analysis of study data, Vascepa 4 g/day statistically significantly reduced ApoC-III levels by 25.1% in MARINE (p < 0.0001) and by 19.2% in ANCHOR (p < 0.0001) versus placebo.
Vascepa was well tolerated in the ANCHOR trial with a safety profile comparable to placebo and there were no treatment-related serious adverse events observed. No significant changes in fasting blood glucose, hemoglobin A1C, vital signs, electrocardiograms, or liver or kidney function were observed with either Vascepa dose. The safety results from the ANCHOR trial are included in the current FDA-approved label for Vascepa.
In April 2015, we received a Complete Response Letter, or CRL, from the FDA in response to our supplemental new drug application, or sNDA, that sought approval of Vascepa for use in patients with mixed dyslipidemia, based on the successful ANCHOR study. The CRL followed an October 2013 rescission by the FDA of a SPA agreement and three failed attempts by us to appeal that rescission at FDA. The FDA has acknowledged the success of the ANCHOR study, which met all primary and secondary endpoints.
9
However, FDA determined that there were insufficient data to conclude that drug-induced changes in serum triglycerides could be recognized by the FDA as a valid surrogate for reducing cardiovascular risk in the ANCHOR population for the purpose of regulatory approval of a drug targeted at a triglyceride-lowering indication in this population. The FDA has acknowledged that the standard of proof required by the FDA for approval of a new drug indication is higher than that generally used to inform patient treatment guidelines and that used by physicians in clinical practice. The FDA did not determine that the drug-induced effects of Vascepa, which go beyond triglyceride-lowering, would not actually reduce cardiovascular risk in this population and the FDA has encouraged us to complete the REDUCE-IT outcomes study. Based on our communications with the FDA, it has been our expectation that submission of final positive results from the REDUCE-IT outcomes study is required for the FDA to consider label expansion for Vascepa.
In May 2015, we and a group of independent physicians filed a lawsuit in federal court to permit us to promote to healthcare professionals the use of Vascepa in patients with mixed dyslipidemia so long as the promotion is truthful and non-misleading. This use reflects recognized medical practice but is not covered by current FDA-approved labeling for the drug. Historically, FDA has considered promotion of drug uses not covered by FDA-approved labeling to be illegal off-label promotion, even if such promotion is truthful and non-misleading. In August 2015, we were granted preliminary relief in the form of a declaratory judgment in this lawsuit. The court declaration permits us to promote to healthcare professionals the FDA-reviewed and agreed effects of Vascepa demonstrated in the ANCHOR clinical trial and presentation of the current state of scientific research related to the potential of Vascepa to reduce the risk of cardiovascular disease including through use of peer-reviewed scientific publications of available data. In August 2015, we began to communicate promotional information beyond the MARINE indication to healthcare professionals in the United States as permitted by this court declaration and in March 2016, the parties obtained court approval of negotiated settlement terms under which the FDA and the U.S. government agreed to be bound by the court’s conclusions from the August 2015 declaration that we may engage in truthful and non-misleading speech promoting the off-label use of Vascepa and that certain statements and disclosures that we proposed to make to healthcare professionals were truthful and non-misleading. While we believe we are now permitted under applicable law to more broadly promote Vascepa, the FDA-approved labeling for Vascepa did not change as a result of this litigation and settlement, and neither government nor other third-party coverage or reimbursement to pay for the off-label use of Vascepa promoted under the court declaration was required.
The REDUCE-IT Study (the completed cardiovascular outcomes study)
The REDUCE-IT study was designed to evaluate the efficacy of Vascepa in reducing major cardiovascular events in an at-risk patient population also receiving statin therapy. REDUCE-IT was a multinational, prospective, randomized, double-blind, placebo-controlled, parallel-group study to evaluate the effectiveness of Vascepa, as an add-on to statin therapy, in reducing first major cardiovascular events in an at-risk patient population compared to statin therapy alone. The control arm of the study was comprised of patients on optimized statin therapy plus placebo. The active arm of the study was comprised of patients on optimized statin therapy plus Vascepa. All subjects enrolled in the study had elevated triglyceride levels and either established coronary heart disease or risk factors for coronary heart disease.
In August 2011, we reached agreement with the FDA on a SPA for the design of the REDUCE-IT (Reduction of Cardiovascular Events with EPA—Intervention Trial) cardiovascular outcomes study. A SPA is an evaluation by the FDA of a protocol with the goal of reaching an agreement that the Phase 3 trial protocol design, clinical endpoints, and statistical analyses are acceptable to support regulatory approval. The FDA agreed that, based on the information we submitted to the agency, the design and planned analysis of the REDUCE-IT study adequately addressed the objectives necessary to support a regulatory submission. A SPA is generally binding upon the FDA unless a substantial scientific issue essential to determining safety or efficacy of the drug is identified after the testing begins.
It is believed that the effects of EPA are not due to a single mode of action, such as triglyceride lowering, but rather to multiple mechanisms working together. Studies in the scientific literature explore potentially beneficial effects of EPA on multiple atherosclerosis processes, including endothelial function, oxidative stress, foam cell formation, inflammation/cytokines, plaque formation/progression, platelet aggregation, thrombus formation, and plaque rupture. With respect to triglyceride levels, our scientific rationale for the REDUCE-IT study was supported by (i) epidemiological data that suggests elevated triglyceride levels correlate with increased cardiovascular disease risk, (ii) genetic data that suggests triglyceride and/or triglyceride-rich lipoproteins (as well as low-density lipoprotein cholesterol (LDL cholesterol), known as bad cholesterol) are independently in the causal pathway for cardiovascular disease and (iii) clinical data that suggest substantial triglyceride reduction in patients with elevated baseline triglyceride levels correlates with reduced cardiovascular risk. The REDUCE-IT study was designed to determine the clinical benefit, if any, of stable EPA therapy in statin-treated patients with elevated triglyceride levels.
In September 2011, we engaged a clinical research organization, or CRO, and began initial trial and clinical site preparation for REDUCE-IT. In December 2011, we announced that the first patient was dosed in the study. In 2016, we completed patient enrollment and randomization of 8,179 individual patients into the REDUCE-IT study. Amarin personnel remained blinded to the efficacy and safety data from the REDUCE-IT study until after the study was completed and the database was locked in 2018.
10
On November 10, 2018, we announced primary results from our REDUCE-IT study as late-breaking clinical results at the 2018 Scientific Sessions of the American Heart Association and the results were concurrently published in The New England Journal of Medicine. REDUCE-IT met its primary endpoint demonstrating a 25% relative risk reduction, or RRR, to a high degree of statistical significance (p<0.001), in first occurrence of major adverse CV events, or MACE, in the intent-to-treat patient population with use of Vascepa 4 grams/day as compared to placebo. Patients qualified to enroll in REDUCE-IT had LDL-C between 41-100 mg/dL (median baseline LDL-C75 mg/dL) controlled by statin therapy and various cardiovascular risk factors including persistent elevated triglycerides, or TG, between 135-499 mg/dL (median baseline 216 mg/dL) and either established cardiovascular disease (secondary prevention cohort) or age 50 or more with diabetes mellitus and at least one other CV risk factor (primary prevention cohort). Approximately 59% of the patients had diabetes at baseline, approximately 71% of the patients had established cardiovascular disease at time of enrollment and approximately 29% were primary prevention subjects at high risk for cardiovascular disease. REDUCE-IT also showed a 26% RRR in its key secondary composite endpoint of cardiovascular death, heart attacks and stroke (p<0.001). We commenced the REDUCE-IT trial in 2011 and have expended more than $300 million to fund its completion.
Number needed to treat, or NNT, was 21 for the first occurrence of MACE in the 5-point primary composite endpoint. The NNT is a statistical concept intended to provide a measurement of the impact of a medicine or therapy by estimating the number of patients that need to be treated in order to have an impact on one person.
An additional seven secondary endpoints were achieved below the key secondary endpoint, in order of sequential statistical testing within the prespecified hierarchy:
|
|
• |
Cardiovascular death or nonfatal heart attack: 25% RRR (p<0.001) |
|
|
• |
Fatal or nonfatal heart attack: 31% RRR (p<0.001) |
|
|
• |
Urgent or emergent revascularization: 35% RRR (p<0.001) |
|
|
• |
Cardiovascular death: 20% RRR (p=0.03) |
|
|
• |
Hospitalization for unstable angina: 32% RRR (p=0.002) |
|
|
• |
Fatal or nonfatal stroke: 28% RRR (p=0.01) |
|
|
• |
Total mortality, nonfatal heart attack or nonfatal stroke: 23% RRR (p<0.001) |
The next prespecified secondary endpoint in the hierarchy was the only such endpoint that did not achieve statistical significance although it trended positively:
|
|
• |
Total mortality, which includes mortality from non-cardiovascular and cardiovascular events: 13% RRR (p=0.09) |
Positive REDUCE-IT results were consistent across various patient subgroups, including female/male, diabetic/non-diabetic and secondary/primary prevention.
Overall adverse event rates in REDUCE-IT were similar across treatment groups and Vascepa was well tolerated with a safety profile generally consistent with clinical experience associated with omega-3 fatty acids and current FDA-approved labeling of such products. The safety results from REDUCE-IT are reviewed in The New England Journal of Medicine publication of REDUCE-IT results.
In the REDUCE-IT trial, cardiovascular benefits appeared not to be influenced significantly by TG levels at baseline (above or below 150 mg/dL baseline range) or as achieved at one year, potentially suggesting mechanisms at work with use of Vascepa that are independent of baseline TG levels or therapy-driven reduction in TG levels. Determining the mechanisms responsible for the benefit shown in REDUCE-IT was not the focus of REDUCE-IT. As summarized from the primary results of REDUCE-IT in The New England Journal of Medicine, potential Vascepa mechanisms of action at work in REDUCE-IT may include TG reduction, anti-thrombotic effects, antiplatelet or anticoagulant effects, membrane-stabilizing effects, effects on stabilization and/or regression of coronary plaque and inflammation reduction, each as supported by earlier stage mechanistic studies.
In addition, in the REDUCE-IT trial, the median change in LDL cholesterol levels from baseline was higher in the placebo group versus the Vascepa group (difference of 5.0 mg/dL; p < 0.001). However, a post hoc analysis of REDUCE-IT data, as published in The New England Journal of Medicine, showed no material difference in each of the primary and key secondary cardiovascular risk composite endpoint event rates for placebo patients that experienced an increase in LDL-C at one year versus those with no change or a decrease, and also suggested a similar relative risk reduction regardless of whether there was an increase in LDL cholesterol level among the patients in the placebo group. Moreover, as the authors of the paper published in The New England Journal of Medicine noted, the relatively small differences in LDL-C levels between the groups would not be likely to explain the 25% lower
11
MACE risk observed with Vascepa and the Japan open-label EPA Lipid Intervention Study, or JELIS, an over 18,000 patient cardiovascular outcomes study in Japan of a highly-pure EPA product similar to Vascepa, previously demonstrated a 19% risk reduction without a mineral oil placebo.
Based on the positive REDUCE-IT results, we have begun promoting REDUCE-IT results to healthcare professionals in the United States based on what we believe is our continuing obligation under our First Amendment settlement to ensure that our promotion of Vascepa remains truthful and non-misleading.
We anticipate continuing to publish additional details of the REDUCE-IT study to address scientific interest beyond the primary results of this study derived from the over 35,000 patient years of study accumulated.
Regulatory Pathway for REDUCE-IT Data
We intend to submit an sNDA to the FDA before the end of March 2019 seeking approval to expand the label for Vascepa based on the effects of Vascepa demonstrated in the REDUCE-IT study. The FDA’s determination of standard or priority review will be made when the sNDA is submitted. At this time, we are planning for a standard review with a PDUFA date which is approximately 10-months after the date of the sNDA submission.
Observed Efficacy of Ethyl-EPA
In Japan, ethyl-EPA is marketed under the product name of Epadel by Mochida Pharmaceutical Co. and is indicated for hyperlipidemia and peripheral vascular disease. In an outcomes study called the Japan EPA Lipid Intervention Study, or JELIS study, which consisted of more than 18,000 patients followed over multiple years, Epadel, when used in conjunction with statins, was shown to reduce cardiovascular events by 19% compared to the use of statins alone. In this study, cardiovascular events decreased by approximately 53% compared to statins alone in the subset of primary prevention patients with triglyceride levels of ≥150 mg/dL (median of 272 mg/dL at entry) and HDL-C <40 mg/dL. Epadel has been approved and available by prescription in Japan for over a decade. In 2013, the Japan Ministry of Health approved Epadel for over-the-counter sales. JELIS provided supportive but not conclusive data that EPA drug therapy may reduce major coronary events. JELIS results cannot be generalized to populations outside of Japan due to limitations in the study’s design. Due to the limitation of JELIS, further study was needed through the REDUCE-IT study to determine the clinical benefit, if any, of EPA therapy in statin-treated patients with elevated triglyceride levels in a patient population beyond that studied in JELIS.
Observed Clinical Safety of Vascepa
In REDUCE-IT, Vascepa was well tolerated with a safety profile generally consistent with clinical experience associated with omega-3 fatty acids and current FDA-approved labeling of such products. Excluding the MACE results described above, overall adverse event rates in REDUCE-IT were similar across the statin plus Vascepa and the statin plus placebo treatment groups. There were no significant differences between treatments in the overall rate of treatment emergent adverse events or serious adverse events leading to withdrawal of study drug. The one serious adverse event occurring at a frequency of >2% was pneumonia which occurred at a numerically higher rate in the statin plus placebo treatment group (2.9%) than in the statin plus Vascepa treatment group (2.6%). Adverse events occurring in 5% or greater of patients and more frequently with Vascepa than placebo were peripheral edema (6.5% Vascepa patients versus 5.0% placebo patients), constipation (5.4% Vascepa patients versus 3.6% placebo patients), and atrial fibrillation (5.3% Vascepa patients versus 3.9% placebo patients). There were numerically more serious adverse events related to bleeding in the statin plus Vascepa treatment group although overall rates were low with no fatal bleeding observed in either group and no significant difference in adjudicated hemorrhagic stroke or serious central nervous system or gastrointestinal bleeding events between treatments.
In the MARINE and ANCHOR trials, patients dosed with Vascepa demonstrated a safety profile similar to placebo. There were no treatment-related serious adverse events in the MARINE study or in the ANCHOR study. In the MARINE and ANCHOR trials, the most commonly reported adverse reaction (incidence >2% and greater than placebo) in Vascepa treated patients was arthralgia (joint pain) (2.3% for Vascepa vs. 1.0% for placebo). There was no reported adverse reaction > 3% and greater than placebo.
Prior to commencing the MARINE, ANCHOR and REDUCE-IT trials, we conducted a pre-clinical program for Vascepa, including toxicology and pharmacology studies. In addition, we previously investigated Vascepa in central nervous system disorders in several double-blind, placebo-controlled studies, including Phase 3 trials in Huntington’s disease. Over 1,000 patients have been dosed with Vascepa in these studies, with over 100 receiving continuous treatment for a year or more. In all studies performed to date, Vascepa has shown a favorable safety and tolerability profile.
In addition to the MARINE and ANCHOR trials, we completed a 28-day pharmacokinetic study in healthy volunteers, a 26-week study to evaluate the toxicity of Vascepa in transgenic mice and multiple pharmacokinetic drug-drug interaction studies in
12
healthy subjects in which we evaluated the effect of Vascepa on certain common prescription drugs. All findings from these studies were consistent with our expectations and confirmed the overall safety profile of Vascepa.
Since Vascepa was made commercially available in 2013, more than five million estimated normalized total prescriptions of Vascepa have been reported by Symphony Health.
New Lipid Compounds and other Preclinical Programs
We are also considering development of other next generation compounds based on our internal lipid science expertise, including potential combination and derivative therapies.
In August 2013, we completed dosing of AMR102, a fixed dose combination of Vascepa and a leading statin product. The study is a randomized, open-label, single-dose, 4-way cross-over study to continue testing of the relative bioavailability of AMR102 capsules, Vascepa capsules with the selected statin taken concomitantly, Vascepa taken alone and the selected statin taken alone. The results of this study support the feasibility of AMR102. We have suspended additional development of AMR102 pending FDA approval of label expansion of Vascepa, anticipated to occur no sooner than after FDA review of the results from the REDUCE-IT study.
We believe that Vascepa and other lipid-based compositions may have an impact on a number of biological factors in the body such as anti-inflammatory mechanisms, cell membrane composition and plasticity, triglyceride levels and regulation of glucose metabolism. Currently all other development activities are at formulation or pre-clinical stages.
Manufacturing and Supply for Vascepa
We manage the manufacturing and supply of Vascepa and have done so since we began clinical development of Vascepa prior to the drug’s marketing approval by the FDA in 2012. We rely on contract manufacturers in each step of our commercial and clinical product supply chain. These steps include active pharmaceutical ingredient, or API, manufacturing, encapsulation of the API, product packaging and supply-related logistics. Our approach to product supply procurement is designed to mitigate risk of supply interruption and maintain an environment of cost competition through diversification of contract manufacturers at each stage of the supply chain and lack of reliance on any single supplier.
The FDA has approved several international large-scale API manufacturers, global encapsulation leaders and two U.S.-based packagers for use in the manufacturing of Vascepa. All of our manufacturing facilities were approved by the FDA following successful preapproval inspections and they remain active manufacturers of Vascepa under FDA authority.
The API material that constitutes ethyl-EPA is a chemical modification of a naturally occurring substance that is derived from specific fish sourced from qualified producers. The fishing from which the raw material for Vascepa is derived is regulated by local government agencies under policies designed to ensure sustainability of the marine life supply. A limited number of other manufacturers have the ability, scale, know-how, sufficient supply chain capability and suitable, industrial-scale facilities to produce ethyl-EPA to the required level of purity. Among the conditions for FDA approval of a pharmaceutical product is the requirement that the manufacturer’s quality control and manufacturing procedures are validated and conform to pharmaceutical current Good Manufacturing Practice, or cGMP, which, under applicable regulations, must be followed at all times. The FDA typically inspects manufacturing facilities before regulatory approval of a product candidate, such as Vascepa, and on a periodic basis after the initial approval. Consistent with cGMP regulations, pharmaceutical manufacturers must expend resources and time to ensure compliance with product specifications as well as production, record keeping, quality control, reporting, and other regulatory requirements.
Some of our agreements with our API suppliers are exclusive and include minimum purchase commitments. During 2018, we fully met the aggregate minimum purchase requirements in our supply agreements. Under the supply agreements, we can purchase more than the minimum requirements. Certain of these agreements contemplate phased capacity expansion aimed at creating sufficient volumes to meet anticipated demand for Vascepa. Certain of these agreements contain provisions for reduced payments (fractional API cost) for unmet annual volume requirements.
We currently market Vascepa in the United States through our direct sales force which grew from approximately 170 sales professionals, including sales representatives and their managers, in September 2018 prior to REDUCE-IT results to approximately 440 sales professionals, including approximately 400 sales representatives, to begin 2019. We currently target clinicians who are top prescribers of lipid-regulating therapies, including statins. During the period from January 2013, when Vascepa was commercially launched in the United States, until October 2013, when the FDA notified us that it rescinded the ANCHOR study SPA agreement, our direct sales force consisted of approximately 275 sales representatives. From early 2014 until September 2018, the size of our direct sales force has included approximately 130 to 150 sales representatives with focus on select sales territories that have the greatest
13
potential for Vascepa sales growth. After topline REDUCE-IT results were announced in September 2018, we began to hire and train additional sales representatives and started 2019 with approximately 400 sales representatives.
From May 2014 through December 2018, in addition to Vascepa promotion by our sales representatives, Kowa Pharmaceuticals America, Inc. co-promoted Vascepa in the United States. This co-promotion reached its mutually agreed upon termination date in December 2018.
We also employ various medical affairs and marketing personnel to support our commercialization of Vascepa. We expanded certain medical education and market awareness initiatives, including, pilot testing of new promotional initiatives following the reporting of positive REDUCE-IT results in 2018 and we intend to further expand such initiatives following label expansion of Vascepa, subject to FDA approval of such expanded label, as discussed below. Our field sales efforts are further complemented by investments in digital and non-personal channels as well as peer-to-peer (e.g., promotional medical education programs & product theaters) initiatives to further increase Vascepa brand awareness and clarify Vascepa’s unique clinical profile.
Since commercial launch of Vascepa in January 2013, we have promoted Vascepa based on the MARINE clinical trial data as reflected in the FDA-approved label for Vascepa. In August 2015, we and our co-promotion partner began communicating promotional information beyond MARINE clinical trial data to targeted healthcare professionals. Such qualified communications are being made pursuant to the August 7, 2015 federal district court declaration and related March 2016 settlement allowing truthful and non-misleading promotion of the FDA-reviewed and agreed effects of Vascepa demonstrated in the ANCHOR clinical trial. This promotion also includes information related to the current state of scientific research about the potential of Vascepa to reduce the risk of cardiovascular disease, including REDUCE-IT data and previously other peer-reviewed scientific publications of available data.
After results of REDUCE-IT were available in September 2018 and demonstrated that Vascepa is effective in lowering the rate of major adverse cardiovascular events in statin-treated patients with CV risk factors, we expanded the size of our U.S. direct sales force and continue to expand promotion of Vascepa based on the results of the REDUCE-IT trial. After publication of the primary results of the REDUCE-IT study in The New England Journal of Medicine and scientific presentation of REDUCE-IT results at the 2018 Scientific Sessions of the American Heart Association (AHA) on November 10, 2018, we updated and expanded our communication of REDUCE-IT results to include the publication and the peer-reviewed information presented in an effort to further ensure that our communications remain truthful and non-misleading.
The dataset from Vascepa is large, representing greater than 35,000 patient years of study. Additionally, the list of prespecified endpoints which we intend to evaluate in support of the sNDA is extensive. We intend to submit an sNDA to the FDA in the United States before the end of March 2019 seeking approval to expand the label for Vascepa based on the effects of Vascepa demonstrated in the REDUCE-IT study. Assuming a standard 10-month review by the FDA, we do not expect an expanded label for Vascepa to be available in 2019 or to impact 2019 Vascepa revenue levels. After the sNDA is submitted, Amarin will seek clarification as to whether priority review by the FDA is possible for this important submission.
Throughout 2019, we expect to continue an efficient expansion of our commercial activities and capabilities directed primarily toward targeted providers and payor decision-makers. We plan to continue to bring the results of REDUCE-IT to healthcare providers and payors, in a manner we believe is both truthful and non-misleading and consistent with our March 2016 settlement. Thus, we are directly connecting Vascepa with the REDUCE-IT data for these target audiences, in advance of having a new label. Further, anticipating the potential receipt of a new, cardiovascular risk reduction indication in the United States, we are preparing a robust, direct to consumer campaign to be launched in earnest after receiving a new label. Concurrently, we also will reassess whether approximately 440 sales professionals, including approximately 400 sales representatives, in conjunction with planned medical education, digital and non-personal outreach levels are adequate and invest appropriately to support the multi-billion-dollar potential of this important new cardiovascular therapy.
Outside of the United States, we have expanded our commercialization activities through partnering arrangements in certain territories. In February 2015, we entered into a Development, Commercialization and Supply Agreement, or the DCS Agreement, with Eddingpharm (Asia) Macao Commercial Offshore Limited, or Eddingpharm, related to the development and commercialization of Vascepa in Mainland China, Hong Kong, Macau and Taiwan, or the China Territory. Under the DCS Agreement, Eddingpharm will be solely responsible for development and commercialization activities in the China Territory and associated expenses. Additionally, Eddingpharm is required to conduct clinical trials in the China Territory to secure regulatory approval in certain territories. For example, in December 2017, Eddingpharm commenced a pivotal clinical trial aimed to support the regulatory approval of the first indication of Vascepa in a patient population with severe hypertriglyceridemia in Mainland China. Additional clinical development efforts may be necessary in this market. Significant commercialization of Vascepa in the China Territory is several years away, if at all. If Eddingpharm is not able to effectively develop and commercialize Vascepa in the China Territory, we may not be able to generate revenue from the DCS Agreement resulting from the sale of Vascepa in the China Territory.
In March 2016, we entered into an agreement with Biologix FZCo, or Biologix, to register and commercialize Vascepa in several Middle Eastern and North African countries. Under the terms of the distribution agreement, we granted to Biologix a non-
14
exclusive license to use our trademarks in connection with the importation, distribution, promotion, marketing and sale of Vascepa in the Middle East and North Africa territory. Commercialization across the Middle East and North Africa is several years away, if at all, in the most commercially significant territories and subject to similar risks as in the China Territory.
In September 2017, we entered into an agreement with HLS Therapeutics Inc., or HLS, to register, commercialize and distribute Vascepa in Canada. Under the agreement, HLS will be responsible for regulatory and commercialization activities and associated costs. We will be responsible for providing assistance towards local filings, supplying finished product under negotiated supply terms, maintaining intellectual property, and continuing the development and funding of REDUCE-IT. Significant commercialization of Vascepa in Canada is years away, if at all. If HLS Therapeutics is not able to effectively register and commercialize Vascepa in Canada, we may not be able to generate revenue from the agreement as a result of the sale of Vascepa in Canada.
We continue to assess other partnership opportunities for licensing Vascepa to partners outside of the United States.
The biotechnology and pharmaceutical industries are highly competitive. There are many pharmaceutical companies, biotechnology companies, public and private universities and research organizations actively engaged in the research and development of products that may be similar to our products. It is probable that the number of companies seeking to develop products and therapies similar to our products will increase. Many of these and other existing or potential competitors have substantially greater financial, technical and human resources than we do and may be better equipped to develop, manufacture and market products. These companies may develop and introduce products and processes competitive with or superior to ours. In addition, other technologies or products may be developed that have an entirely different approach or means of accomplishing the intended purposes of our products, which might render our technology and products noncompetitive or obsolete.
Our competitors both in the United States and abroad include large, well-established pharmaceutical and generic companies, specialty and generic pharmaceutical sales and marketing companies, and specialized cardiovascular treatment companies. GlaxoSmithKline plc currently sells Lovaza®, a prescription-only omega-3 fatty acid indicated for patients with severe hypertriglyceridemia, which was approved by FDA in 2004 and has been on the market in the United States since 2005. Multiple generic versions of Lovaza are available in the United States. Other large companies with competitive products include AbbVie, Inc., which currently sells Tricor® and Trilipix® for the treatment of severe hypertriglyceridemia and Niaspan®, which is primarily used to raise high-density lipoprotein cholesterol, or HDL-C, but is also used to lower triglycerides. Multiple generic versions of Tricor, Trilipix and Niaspan are also available in the United States. We compete with these drugs, and in particular, multiple low-cost generic versions of these drugs, in our FDA-approved indicated use and in off-label uses, such as to beneficially affect lipid levels in patients with persistent high triglyceride levels after statin therapy with the aim of potentially lowering cardiovascular risk beyond statin therapy.
In addition, in May 2014, Epanova® (omega-3-carboxylic acids) capsules, a free fatty acid form of omega-3 (comprised of 55% EPA and 20% DHA), was approved by the FDA for patients with severe hypertriglyceridemia. Epanova was developed by Omthera Pharmaceuticals, Inc., and is now owned by AstraZeneca Pharmaceuticals LP (AstraZeneca). Also, in April 2014, Omtryg, another omega-3-acid fatty acid composition developed by Trygg Pharma AS, received FDA approval for severe hypertriglyceridemia. Neither Epanova nor Omtryg have been commercially launched, but could launch at any time. Each of these competitors, other than Trygg, has greater resources than we do, including financial, product development, marketing, personnel and other resources. Neither Epanova nor Omtryg have been commercially launched.
AstraZeneca is currently conducting a long-term outcomes study to assess Statin Residual Risk Reduction With EpaNova in HiGh Cardiovascular Risk PatienTs With Hypertriglyceridemia (STRENGTH). The study is a randomized, double-blind, placebo-controlled (corn oil), parallel group design that is believed to have enrolled approximately 13,000 patients with hypertriglyceridemia and low HDL and high risk for cardiovascular disease randomized 1:1 to either corn oil plus statin or Epanova plus statin, once daily, for approximately 3-5 years as determined when the number of major adverse cardiovascular event outcomes is reached. The STRENGTH study is estimated to be completed in 2020, but it could be stopped earlier if, for example, it generates an overwhelming efficacy result. In addition, Kowa Research Institute (a subsidiary of the Japanese company Kowa Co., Ltd) announced in March 2017 that it is initiating a phase III cardiovascular outcomes trial titled PROMINENT examining the effect of pemafibrate (experimental name K-877) in reducing cardiovascular events in Type II diabetic patients with hypertriglyceridemia. Kowa Research Institute has publicly estimated study completion in May 2022, and if successful, U.S. regulatory approval is estimated in mid-2023.
During 2018, two outcomes studies were completed of omega-3 mixtures which both failed to achieve their primary endpoints of cardiovascular risk reduction and two meta-analyses were published showing that omega-3 mixtures are not effective in lowering cardiovascular risk. Results of these failed outcomes studies and analysis, while not done with Vascepa, may negatively affect sales of Vascepa. For example, results of VITamin D and OmegA-3 TriaL (VITAL), as announced immediately before the presentation of REDUCE-IT results at the 2018 Scientific Sessions of the American Heart Association (AHA) on November 10, 2018, failed to achieve its primary endpoint of lowering cardiovascular events. VITAL was an NIH funded randomized double-blind, placebo-controlled, 2x2 factorial trial of 2000 IU per day of vitamin D3 and 1 gram per day of omega-3 fatty acid supplementation (Lovaza)
15
for the primary prevention of cancer and cardiovascular disease in a nationwide USA cohort of 25,874 adults not selected for elevated cardiovascular or cancer risk.
Likewise, in 2018, results from A Study of Cardiovascular Events iN Diabetes (ASCEND) trial were released and showed negligible results for omega-3 fatty acids 1 gram daily. ASCEND was a British Heart Foundation funded 2x2 factorial design, randomized study to assess whether aspirin 100 mg daily versus placebo and separately, omega-3 fatty acids 1 gram daily versus placebo, reduce the risk of cardiovascular events in a nationwide UK cohort of over 15,000 individuals with diabetes who do not have atherosclerotic cardiovascular disease. In addition, VITAL showed that supplementation with either omega-3 fatty acid at a dose of 1 gram per day or vitamin D3 at a dose of 2000 IU per day was not effective for primary prevention of CV or cancer events among healthy middle-aged men and women across 5 years of follow up.
In meta-analysis, presented in 2018 by the Cochran Foundation and separately as published in JAMA, additional omega-3 studies were evaluated. Similar to the VITAL and ASCEND studies, most of the studies in these omega-3 meta-analyses were of omega-3 mixtures, including DHA, and most were studies of relatively low doses of omega-3 as is associated with dietary supplementation and/or they studied relatively low risk patient populations. The exception was the JELIS study, conducted in Japan, of highly pure EPA which demonstrated a positive outcome benefit. The negative results from such omega-3 mixture studies could create misleading impressions about the use of omega-3s generally, including Vascepa, despite REDUCE-IT positive results and the highly-pure and stable EPA active ingredient in Vascepa and its higher dose regimen.
We are also aware of other pharmaceutical companies that are developing products that, if successfully developed, approved and marketed, would compete with Vascepa. Acasti Pharma, or Acasti, a subsidiary of Neptune Technologies & Bioresources Inc., announced in December 2015 that it intends to pursue a regulatory pathway under section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act, or FDCA, for its omega-3 prescription drug candidate, CaPre® (omega-3 phospholipid), derived from krill oil, for the treatment of hypertriglyceridemia. In September 2016, Acasti announced positive results from its pivotal bioavailability bridging study comparing CaPre to Lovaza, establishing a scientific bridge between the two that is expected to support the feasibility of a 505(b)(2) regulatory pathway. Acasti initiated a Phase 3 clinical program (TRILOGY) to assess the safety and efficacy of CaPre in patients with very high (≥500 mg/dL) triglycerides in the first quarter of 2018. Acasti completed enrollment in Q4 2018 and study completion is expected by the end of 2019. We believe Micelle BioPharma Inc., or Micelle, is also developing potential treatments for hypertriglyceridemia based on omega-3 fatty acids. To our knowledge, Micelle, after acquiring SC401 from Sancilio & Company, or Sancilio, is pursuing a regulatory pathway under section 505(b)(2) of the FDCA for its product and submitted an Investigational New Drug Application, or IND, in July 2015. Micelle (Sancilio) completed two pharmacokinetic studies and Phase 2 bioavailability studies (FASTR I&II), with one comparing SC401 to Lovaza. We expect the company or a potential partner to initiate a pivotal clinical Phase 3 study as the next step in development.
Matinas BioPharma, Inc. is developing an omega-3-based therapeutic (MAT9001) for the treatment of severe hypertriglyceridemia and mixed dyslipidemia. In the fourth quarter of 2014, Matinas BioPharma, Inc. filed an IND with the FDA to conduct a human study in the treatment of severe hypertriglyceridemia and, in June 2015, the company announced topline results for its head-to-head comparative short duration pharmacokinetic and pharmacodynamic study of MAT9001 versus Vascepa in patients under conditions inconsistent with the FDA-approved label for Vascepa and presented results based on biomarker modification without outcomes data. In September 2017, Matinas announced that it will be seeking a partner company to develop and commercialize MAT9001. Akcea Therapeutics/Ionis Pharmaceuticals (formerly Isis Pharmaceuticals), or Akcea/Ionis, in August 2018 announced the receipt of a complete response letter from the FDA for WAYLIVRA™ (volanesorsen), a drug candidate administered through weekly subcutaneous injections, for the treatment of familial chylomicronemia syndrome (FCS). Akcea will continue to work with the FDA on the path forward for Waylivra for the treatment of FCS. Waylivra continues to be developed for the treatment of familial partial lipodystrophy (FPL).
A Phase 3 trial is currently ongoing studying Waylivra (volanesorsen) in patients with FPL (BROADEN trial). Akcea/Ionis expects to file an NDA for FPL in 2019. In January 2017, Akcea/Ionis announced a strategic collaboration and option agreement with Novartis whereby Novartis will help develop (including funding cardiovascular outcomes studies) and commercialize products emerging from this collaboration, including Waylivra (volanesorsen). In June 2018, Gemphire Therapeutics announced positive topline results from a Phase 2b trial (INDIGO-1) of its drug candidate, gemcabene, in patients with severe hypertriglyceridemia. Gemcabene is an oral, once-daily pill for a number of hypercholesterolemic populations and severe hypertriglyceridemia. Gemphire announced plans to initiate a Phase III study for homozygous familial hypercholesterolemia (HoFH), heterozygous familial hypercholesterolemia (HeFH) and non-familial hypercholesterolemia in ASCVD patients in the second half of 2018. In August 2018, the FDA requested that Gemphire conduct an additional long-term toxicity study before commencing any further clinical testing, thereby effectively placing gemcabene on clinical hold. Zydus Cadila has a Phase 2 development program for its lead molecule, Saroglitazar, in various indications, including severe hypertriglyceridemia in the United States. In August 2018, the Company announced that it had suspended the Phase 2 trial in the severe hypertriglyceridemia indication due to study enrollment issues, while it continues development activities in other indications. The product is approved in India under the name Lipaglyn® for the treatment of hypertriglyceridemia and diabetic dyslipidemia. We are also aware that bezafibrate has been licensed by Intercept Pharmaceuticals to be further developed and potentially launched in the United States market.
16
Based on prior communications from the FDA, including communications in connection with its review of the ANCHOR indication for Vascepa, it is our understanding that the FDA is not prepared to approve any therapy for treatment of cardiovascular risk based on biomarker modification without outcomes study data, with the potential exception of therapies which lower LDL-cholesterol. In particular, it is our understanding that the FDA is not prepared to approve any therapy based on data demonstrating lowering of triglyceride levels. In our view, this position from the FDA is unlikely to change based on the REDUCE-IT study particularly in light of the independence of the positive benefit demonstrated in the REDUCE-IT study from triglyceride levels and benefit from the REDUCE-IT study supporting that the positive effects of Vascepa are unique to Vascepa and extend beyond triglyceride reduction. If the FDA were to change this position, it could potentially have a negative impact on Amarin by making it easier for other products to achieve a cardiovascular risk reduction indication without the need in advance to conduct a long and expensive cardiovascular outcomes study
Vascepa also faces competition from dietary supplement companies marketing omega-3 products as nutritional supplements. Such products are classified as food, not as prescription drugs or as over-the-counter drugs, by the FDA. Many of the promoters of such products have greater resources than Amarin and they are not restricted to the same standards as are prescription drugs with respect to promotional claims or manufacturing quality, consistency and subsequent product stability. Though we have taken legal action against supplement manufacturers attempting to use the REDUCE-IT results to promote their products, we cannot be sure physicians and pharmacists will view the FDA-approved prescription-only status, EPA-only purity of Vascepa and stringent regulatory oversight as significant advantages versus omega-3 dietary supplements regardless of clinical study results and other scientific data.
In addition, several generic drug companies have sought to challenge the validity and enforceability of our patents and have submitted to FDA applications for approval of generic versions of Vascepa.
Government Regulation and Regulatory Matters
Any product development activities related to Vascepa or products that we may develop or acquire in the future will be subject to extensive regulation by various government authorities, including the FDA and comparable regulatory authorities in other countries, which regulate the design, research, clinical and non-clinical development, testing, manufacturing, storage, distribution, import, export, labeling, advertising and marketing of pharmaceutical products and devices. Generally, before a new drug can be sold, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific to each regulatory authority, submitted for review and approved by the regulatory authority. The data is generated in two distinct development stages: pre-clinical and clinical. Drugs must be approved by the FDA through the NDA process before they are first marketed in the United States. For new chemical entities, the pre-clinical development stage generally involves synthesizing the active component, developing the formulation, determining the manufacturing process and controls, as well as carrying out non-human toxicology, pharmacology and drug metabolism studies which support subsequent clinical testing.
The clinical stage of development can generally be divided into Phase 1, Phase 2 and Phase 3 clinical trials. In Phase 1, generally, a small number of healthy volunteers are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these studies is to assess the metabolism, pharmacologic action, side effect tolerability and safety of the drug. Phase 2 trials typically involve studies in disease-affected patients to determine the dose required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected. Phase 3 trials generally involve large numbers of patients at multiple sites, in multiple countries and are designed to provide the pivotal data necessary to demonstrate the effectiveness of the product for its intended use, its safety in use, and may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing.
United States Drug Development
In the United States, the process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Prior to the start of human clinical studies for a new drug in the United States, preclinical laboratory and animal tests are often performed under the FDA’s Good Laboratory Practices regulations, or GLP, and an investigational new drug application, or IND, is filed with the FDA. Similar filings are required in other countries; however, data requirements and other information needed for a complete submission may differ in other countries. The amount of data that must be supplied in the IND depends on the phase of the
17
study. Phase 1 studies typically require less data than larger Phase 3 studies. A clinical plan must be submitted to the FDA prior to commencement of a clinical trial. If the FDA has concerns about the clinical plan or the safety of the proposed studies, it may suspend or terminate the study at any time. Studies must be conducted in accordance with Good Clinical Practice, or GCP, and regular reporting of study progress and any adverse experiences is required. Studies are also subject to review by independent institutional review boards, or IRBs, responsible for overseeing studies at particular sites and protecting human research study subjects. An independent IRB may also suspend or terminate a study once initiated.
NDA and FDA Review Process
Following trial completion, trial data is analyzed to determine safety and efficacy. Data is then filed with the FDA in an NDA along with proposed labeling for the product and information about the manufacturing and testing processes and facilities that will be used to ensure product quality. The NDA must contain proof of safety, purity, potency and efficacy, which entails extensive pre-clinical and clinical testing. FDA approval of an NDA must be obtained before first marketing of a drug in the United States.
The FDA will likely re-analyze the clinical trial data, which could result in iterative discussions between the FDA and us during the review process. The review and evaluation of applications by the FDA is extensive and time consuming and may take longer than originally planned to complete. The FDA may conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with current Good Manufacturing Practice, or cGMP, requirements and may also audit data from clinical and pre-clinical trials.
There is no assurance that the FDA will ultimately approve a drug product for marketing in the United States. Even if future indications for Vascepa are approved, the FDA’s review will be lengthy, and we may encounter significant difficulties or costs during the review process. After approving any drug product, the FDA may require post-marketing testing and surveillance to monitor the effects of approved products or it may place conditions on approvals including potential requirements or risk management plans that could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following initial marketing.
Off-label Promotion in the United States
The Federal Food, Drug, and Cosmetic Act, or FDCA, has been interpreted by the FDA and the U.S. government to make it illegal for pharmaceutical companies to promote their FDA-approved products for uses that have not been approved by the FDA. Companies that market drugs for off-label uses or indications have been subject to related costly litigation, criminal penalties and civil liability under the FDCA and the False Claims Act. However, recent case law has called into question the extent to which government in the United States, including FDA, can, and is willing to seek to, prevent truthful and non-misleading speech related to off-label uses of FDA-approved products such as Vascepa.
In May 2015, we and a group of independent physicians filed a lawsuit against the FDA seeking a federal court declaration that would permit us and our agents to promote to healthcare professionals the use of Vascepa in the ANCHOR population and promote on the potential of Vascepa to reduce the risk of cardiovascular disease so long as the promotion is truthful and non-misleading. This use of Vascepa at issue reflected recognized medical practice but was not approved by the FDA and is thus not covered by current FDA-approved labeling for the drug. Promotion of an off-label use has generally been considered by the FDA to be illegal under the FDCA. The lawsuit, captioned Amarin Pharma, Inc., et al. v. Food & Drug Administration, et al., 119 F. Supp. 3d 196 (S.D.N.Y. 2015), was filed in the United States District Court for the Southern District of New York. In the lawsuit, we contended principally that FDA regulations limiting off-label promotion of truthful and non-misleading information are unconstitutional under the freedom of speech clause of the First Amendment to the U.S. Constitution as applied in the case of our proposed promotion of Vascepa. The physicians in the suit regularly treated patients at risk of cardiovascular disease and, as the complaint contended, have First Amendment rights to receive truthful and non-misleading information from Amarin. The suit was based on the principle that better informed physicians make better treatment decisions for their patients. The FDA opposed this lawsuit but did not dispute the veracity of the subject ANCHOR clinical trial data (the safety data from which was already and currently is in FDA-approved labeling of Vascepa) or the peer-reviewed research related to Vascepa and the potential for cardiovascular risk reduction.
In connection with this litigation, the FDA sent a detailed letter to us on June 5, 2015 that confirmed the validity of the ANCHOR trial results. The letter also sought to clarify how, in the FDA’s view, applicable law and FDA policies apply to the communications proposed in our complaint. The FDA stated in this letter that it did not have concerns with much of the information we proposed to communicate and provided us with guidance on the FDA’s view of lawful, but limited paths for the dissemination and communication to healthcare professionals of the effects of Vascepa demonstrated in the ANCHOR clinical trial and use of peer-reviewed scientific publications in the context of appropriate disclaimers.
In August 2015, we were granted preliminary relief in this lawsuit through the court’s declaratory judgment that confirmed we may engage in truthful and non-misleading speech promoting the off-label use of Vascepa to healthcare professionals, i.e., to treat patients with persistently high triglycerides, and that such speech may not form the basis of a misbranding action under the FDCA. In
18
August 2015, we began to communicate promotional information beyond the MARINE indication to healthcare professionals in the United States as permitted by this court declaration. The FDA did not appeal the court’s ruling.
In March 2016, we settled this litigation under terms by which the FDA and the U.S. government agreed to be bound by the conclusions from the federal court order that we may engage in truthful and non-misleading speech promoting the off-label use of Vascepa and that certain statements and disclosures that we proposed to make to healthcare professionals were truthful and non-misleading. As part of the settlement, given, as expressed in the court’s opinion, that the dynamic nature of science and medicine is that knowledge is ever-advancing and that a statement that is fair and balanced one day may become incomplete or otherwise misleading in the future as new studies are done and new data is acquired, we agreed that we bear the responsibility to ensure that our communications regarding off-label use of Vascepa remain truthful and non-misleading, consistent with the federal court ruling.
While we believe we are now permitted under our settlement and the freedom of speech clause of the First Amendment to the United States Constitution to more broadly promote Vascepa, the FDA-approved labeling for Vascepa did not change as a result of this litigation and settlement. In addition, under our settlement, neither government nor other third-party coverage or reimbursement to pay for the off-label use of Vascepa was required. Based on our communications with the FDA, we expect that the FDA’s review and analysis of our final positive results from the REDUCE-IT outcomes study will be required for FDA-approved label expansion for Vascepa. However, we proactively communicate results from the REDUCE-IT trial in a manner we believe is truthful and non-misleading and thus protected under the freedom of speech clause of the First Amendment to the United States Constitution.
Even though we have the benefit of a final settlement in this litigation, our promotion is still subject to a high, perhaps abnormally high, degree of scrutiny to ensure that our promotion remains within the scope covered by the settlement. For example, under the settlement, we remain responsible for ensuring our speech is truthful and non-misleading. Data arising from studies of drug products are complex, such as the many studies that we believe show supportive but not conclusive research on the potential connection between the effects of EPA, the active ingredient in Vascepa, and cardiovascular risk reduction (e.g., the JELIS trial of a highly-pure EPA product in Japan by Mochida Pharmaceutical Co., Ltd., or Mochida, and other data using a variety of levels of evidence that connect EPA to favorable effects toward reduced cardiovascular risk). We, the FDA, the U.S. government, our competitors and other interested parties may not agree on the truthfulness and non-misleading nature of our promotional materials with respect to the outcome of these trials or other direct or indirect claims we make about Vascepa. Likewise, the FDA, the U.S. government, our competitors and other interested parties may not agree on the truthfulness and non-misleading nature of our promotional materials related to the REDUCE-IT results. Federal and state governments or agencies may also seek to find other means to prevent our promotion of unapproved truthful and non-misleading information about Vascepa. If our promotional activities or other operations are found to be in violation of any law or governmental regulation through existing or new interpretations, we may be subject to prolonged litigation, penalties, including civil and criminal penalties, damages, fines and the curtailment or restructuring of our operations. Also, if governmental parties or our competitors view our claims as misleading or false, we could also be subject to liability based on fair competition-based statutes, such as the Lanham Act. Any of such negative circumstances could adversely affect our ability to operate our business and our results of operations.
Foreign Regulation of New Drug Compounds
In addition to regulations in the United States, we may be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in all or most foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application, or CTA, much like the IND prior to the commencement of human clinical trials. In Europe, for example, a CTA must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed. Similarly, clinical trials conducted in countries such as Australia, Canada, and New Zealand, require review and approval of clinical trial proposals by an ethics committee, which provides a combined ethical and scientific review process. Most countries in which clinical studies are conducted require the approval of the clinical trial proposals by both the regulatory body and ethics committee.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials must be conducted in accordance with GCP, which have their origin in the World Medical Association’s Declaration of Helsinki, the applicable regulatory requirements, and guidelines developed by the International Conference on Harmonization, or ICH, for GCP practices in clinical trials.
Post-Marketing Requirements
Following approval of a new product, a pharmaceutical company generally must engage in numerous specific monitoring and recordkeeping activities, such as routine safety surveillance, and must continue to submit periodic and other reports to the applicable
19
regulatory agencies, including any cases of adverse events and appropriate quality control records. Such reports submitted to the FDA may result in changes to the label and/or other post-marketing requirements or actions, including product withdrawal. These are viable risks once a product is on the market. Additionally, modifications or enhancements to the products or labeling or changes of site of manufacture are often subject to the approval of the FDA and other regulators, which may or may not be received or may result in a lengthy review process.
Prescription drug advertising is subject to federal, state and foreign regulations. In the United States, the FDA regulates prescription drug promotion, including direct-to-consumer advertising. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use. Any distribution of prescription drug products and pharmaceutical samples must comply with the U.S. Prescription Drug Marketing Act, or the PDMA, a part of the FDCA.
In the United States, once a product is approved, its manufacture is subject to comprehensive and continuing regulation by the FDA. The FDA regulations require that products be manufactured in specific approved facilities and in accordance with pharmaceutical cGMPs, and NDA holders must list their products and register their manufacturing establishments with the FDA. These regulations also impose certain organizational, procedural and documentation requirements with respect to manufacturing and quality assurance activities. NDA holders using contract manufacturers, laboratories or packagers are responsible for the selection and monitoring of qualified firms, and, in certain circumstances, qualified suppliers to these firms. These firms and, where applicable, their suppliers are subject to inspections by the FDA at any time, and the discovery of violative conditions, including failure to conform to cGMPs, could result in enforcement actions that interrupt the operation of any such facilities or the ability to distribute products manufactured, processed or tested by them.
Federal and State Fraud and Abuse Laws
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws restrict certain marketing practices in the biopharmaceutical industry. These laws include anti-kickback statutes and false claims statutes.
The federal anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, or receiving remuneration to induce or in return for a referral or the purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any healthcare facility, item or service reimbursable under Medicare, Medicaid, or other federal healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. Liability may be established without a person or entity having actual knowledge of the federal anti-kickback statute or specific intent to violate it. In addition, the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases, or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection from anti-kickback liability. Moreover, there are no safe harbors for many common practices, such as educational and research grants or patient or product support programs.
The federal civil False Claims Act prohibits, among other things, any person from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds, or knowingly making or using, or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly concealing, or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. The False Claims Act also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the statute and to share in any monetary recovery. Recently, several pharmaceutical and other healthcare companies have been investigated or faced enforcement actions under the federal civil False Claims Act for a variety of alleged improper marketing activities, including allegations that they caused false claims to be submitted because of the company’s marketing of the product for unapproved, and thus allegedly non-reimbursable, uses. Federal enforcement agencies also have showed increased interest in pharmaceutical companies’ product and patient assistance programs, including reimbursement and co-pay support services, and a number of investigations into these programs have resulted in significant civil and criminal settlements. Pharmaceutical and other healthcare companies also are subject to other federal false claims laws, including, among others, federal criminal healthcare fraud and false statement statutes that extend to non-government health benefit programs.
The Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act (HITECH), collectively referred to herein as HIPAA, among other things, imposes criminal and civil liability for knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payor and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. In addition, HITECH imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information.
20
The federal Physician Payment Sunshine Act, being implemented as the Open Payments Program, which requires manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to direct or indirect payments and other transfers of value to physicians and teaching hospitals, as well as ownership and investment interests held in the company by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report information regarding payments and transfers of value provided to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse-midwives.
The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers. Other states or localities may have laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; restrict the ability of manufacturers to offer co-pay support to patients for certain prescription drugs; require drug manufacturers to report information related to clinical trials, or information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and/or require identification or licensing of sales representatives.
Because of the breadth of these laws and the narrowness of the exceptions or safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of such laws. Such a challenge could have a material adverse effect on our business, financial condition and results of operations. These laws may impact, among other things, our proposed sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business.
If our promotional activities or other operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us through existing or new interpretations, we may be subject to prolonged litigation, penalties, including civil and criminal penalties, damages, fines and the curtailment or restructuring of our operations. Also, if governmental parties or our competitors view our claims as misleading or false, we could also be subject to liability based on fair competition-based statutes, such as the Lanham Act. Any of such negative circumstances could adversely affect our ability to operate our business and our results of operations.
In the United States and foreign jurisdictions, there have been a number of legislative and regulatory changes to the healthcare system that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the United States federal and state levels that seek to reduce healthcare costs. The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for our products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payers often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payers. In addition, there has been renewed interest in amending the Social Security Act to allow Medicare to negotiate prices for prescription drugs covered under Medicare Part B. If this were to be enacted by Congress and signed by the President, the prices we obtain for our products covered under Part B could be lower than the prices we might otherwise obtain, and it could exert a similar lowering pressure on payments from non-governmental payers.
The Agency for Healthcare Research and Quality (AHRQ), established by the MMA and provided additional funding by The American Recovery and Reinvestment Act of 2009, conducts comparative effectiveness research on different treatments for the same illness. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payers, is it possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidates. If third-party payers do not consider our products to be cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
21
In March 2010 the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively the ACA, was enacted, which substantially changed the way healthcare is financed by both governmental and private insurers and significantly impacts the pharmaceutical industry. Among the provisions of the ACA of greatest importance to the pharmaceutical and biotechnology industry are the following:
|
|
• |
an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic products, apportioned among these entities according to their market share in certain government healthcare programs, that began in 2011; |
|
|
• |
a licensure framework for follow-on biologic products; |
|
|
• |
a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and |
|
|
• |
establishment of a Center for Medicare and Medicaid Innovation at the Centers for Medicare & Medicaid Services to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending that began on January 1, 2011. |
Some of the provisions of the ACA have yet to be fully implemented, and certain provisions have been subject to judicial and Congressional challenges. In addition, there have been efforts by the Trump administration to repeal or replace certain aspects of the ACA and to alter the implementation of the ACA and related laws. For example, the Tax Cuts and Jobs Act enacted on December 22, 2017, eliminated the tax-based shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code of 1986, commonly referred to as the “individual mandate,” effective January 1, 2019. Further, the Bipartisan Budget Act of 2018 among other things, amended the Medicare statute, effective January 1, 2019, to reduce the coverage gap in most Medicare drug plans, commonly known as the “donut hole,” by raising the manufacturer discount under the Medicare Part D coverage gap discount program to 70%. It is unclear how the ACA and its implementation, as well as efforts to repeal or replace, or invalidate, the ACA, or portions thereof, will affect our business. It is possible that the ACA will continue to exert pressure on pharmaceutical pricing, especially under the Medicare and Medicaid programs, and may also increase our regulatory burdens and operating costs. Additional legislative changes, regulatory changes, and judicial challenges related to the ACA remain possible.
Pharmaceutical Pricing and Reimbursement
Our ability to successfully commercialize our product depends in significant part on the availability of adequate financial coverage and reimbursement from third-party payors, including, in the United States, governmental payors such as Medicare and Medicaid, as well as managed care organizations, private health insurers and other organizations. Third-party payors decide which drugs they will pay for and establish reimbursement and copayment levels. Third-party payors are increasingly challenging the prices charged for medicines and examining their cost effectiveness, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost effectiveness of our products. Even with studies, our products may be considered less safe, less effective or less cost effective than other products, and third-party payors may not provide coverage and reimbursement for our product candidates, in whole or in part.
Political, economic and regulatory influences are subjecting the healthcare industry in the United States to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could impact our ability to sell our products profitably. We anticipate that the United States Congress, state legislatures and the private sector will continue to consider and may adopt healthcare policies intended to curb rising healthcare costs. These cost containment measures include: controls on government funded reimbursement for drugs; new or increased requirements to pay prescription drug rebates to government healthcare programs; controls on healthcare providers; challenges to the pricing of drugs or limits or prohibitions on reimbursement for specific products through other means; requirements to try less expensive products or generics before a more expensive branded product; changes in drug importation laws; expansion of use of managed care systems in which healthcare providers contract to provide comprehensive healthcare for a fixed cost per person; and public funding for cost effectiveness research, which may be used by government and private third-party payors to make coverage and payment decisions. Further, federal budgetary concerns could result in the implementation of significant federal spending cuts, including cuts in Medicare and other health related spending in the near term. For example, beginning April 1, 2013, Medicare payments for all items and services, including drugs and biologics, were reduced by 2% under the sequestration (i.e., automatic spending reductions) required law and this reduction runs to 2027. These cuts reduce reimbursement payments related to our products, which could potentially negatively impact our revenue.
Payors also are increasingly considering new metrics as the basis for reimbursement rates, such as average sales price, average manufacturer price and Actual Acquisition Cost. CMS surveys and publishes retail community pharmacy acquisition cost information in the form of National Average Drug Acquisition Cost files to provide state Medicaid agencies with a basis of comparison for their own reimbursement and pricing methodologies and rates. It is difficult to project the impact of these evolving reimbursement
22
mechanics on the willingness of payors to cover our products. We participate in the Medicaid Drug Rebate program, the 340B drug pricing program, and the U.S. Department of Veterans Affairs, or VA, Federal Supply Schedule, or FSS, pricing program. Under the Medicaid Drug Rebate program, we are required to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under Medicaid and Part B of the Medicare program.
Federal law requires that any company that participates in the Medicaid Drug Rebate program also participate in the 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B program requires participating manufacturers to agree to charge statutorily defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and Medicaid rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate program.
In order to be eligible to have our products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, we participate in the VA/FSS pricing program. Under this program, we are obligated to make our products available for procurement on an FSS contract and charge a price to four federal agencies - the VA, U.S. Department of Defense, Public Health Service and U.S. Coast Guard - that is no higher than the statutory Federal Ceiling Price, or FCP. The FCP is based on the non-federal average manufacturer price, or Non-FAMP, which we calculate and report to the VA on a quarterly and annual basis. We also participate in the Tricare Retail Pharmacy program, under which we pay quarterly rebates on utilization of innovator products that are dispensed through the Tricare Retail Pharmacy network to Tricare beneficiaries. The rebates are calculated as the difference between the annual Non-FAMP and FCP.
The Medicaid Drug Rebate program, 340B program, and VA/FSS pricing program, and the risks relating to price reporting and other obligations under these programs, are further discussed under the heading “If we fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate program or other governmental pricing programs, we could be subject to additional reimbursement requirements, penalties, sanctions and fines, which could have a material adverse effect on our business, financial condition, results of operations and growth prospects” in Part I, Item 1A of this Annual Report on Form 10-K.
FDA Marketing Exclusivity and Generic Competition
The FDCA, as amended by the Drug Price Competition and Patent Term Restoration Act of 1984, as amended, or the Hatch-Waxman Amendments, provides for market exclusivity provisions that can help protect the exclusivity of new drugs by delaying the acceptance and final approval of certain competitive drug applications. NCE marketing exclusivity precludes approval during the five-year exclusivity period of certain 505(b)(2) applications and abbreviated new drug applications, or ANDAs, submitted by another company for another version of the drug. The timelines and conditions under the ANDA process that permit the start of patent litigation and allow the FDA to approve generic versions of brand name drugs like Vascepa differ based on whether a drug receives three-year, or five-year, NCE marketing exclusivity. In May 2016, after significant litigation, FDA determined that Vascepa is eligible for NCE marketing exclusivity. Accordingly, we believe a related 30-month stay is currently in place with respect to our 1-gram dose strength of Vascepa that is scheduled to continue until January 26, 2020, seven-and-a-half years from FDA approval of Vascepa, unless related patent litigation is resolved against us sooner.
The FDA typically makes a determination on marketing exclusivity in connection with an NDA approval of a drug for a new indication. FDA marketing exclusivity is separate from, and in addition to, patent protection, trade secrets and manufacturing barriers to entry which could also help protect Vascepa against generic competition.
We applied to the FDA for five-year, NCE marketing exclusivity for Vascepa in connection with the NDA for our MARINE indication, which NDA was approved by the FDA on July 26, 2012. On February 21, 2014, in connection with the July 26, 2012 approval of the MARINE indication, the FDA denied a grant of five-year NCE marketing exclusivity to Vascepa and granted three-year marketing exclusivity. Under applicable regulations, such three-year exclusivity would have extended through July 25, 2015 and would have been supplemented by a 30-month stay triggered by patent litigation that would have extended into September 2016, unless such patent litigation was resolved against us sooner.
NCE marketing exclusivity precludes approval during the five-year exclusivity period of certain 505(b)(2) applications and ANDAs submitted by another company for another version of the drug. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. In such case, the pioneer drug company is afforded the benefit of a 30-month stay against the launch of such a competitive product that extends from the end of the five-year exclusivity period. A pioneer company could also be afforded extensions to the stay under applicable regulations, including a six-month pediatric exclusivity extension or a judicial extension if applicable requirements are met. A drug sponsor could also gain a form of marketing
23
exclusivity under the Hatch-Waxman Amendments if such company can, under certain circumstances, complete a human clinical trial process and obtain regulatory approval of its product.
In contrast, a three-year period of exclusivity under the Hatch-Waxman Amendments is generally granted for a drug product that contains an active moiety that has been previously approved, such as when the application contains reports of new clinical investigations (other than bioavailability studies) conducted by the sponsor that were essential to approval of the application. Accordingly, we expect to receive three-year exclusivity in connection with any future regulatory approvals of Vascepa, such as an approval sought based on positive REDUCE-IT outcomes study results. Such three-year exclusivity protection precludes the FDA from approving a marketing application for an ANDA, a product candidate that the FDA views as having the same conditions of approval as Vascepa (for example, the same indication and/or other conditions of use), or a 505(b)(2) NDA submitted to the FDA with Vascepa as the reference product, for a period of three years from the date of FDA approval. The FDA may accept and commence review of such applications during the three-year exclusivity period. Such three-year exclusivity grant does not prevent a company from challenging the validity of patents at any time, subject to any prior four-year period pending from a grant of five-year exclusivity. This three-year form of exclusivity may also not prevent the FDA from approving an NDA that relies only on its own data to support the change or innovation.
On February 27, 2014, we sued the FDA in the U.S. District Court for the District of Columbia to challenge the agency’s denial of five-year NCE exclusivity for Vascepa, based on our reading of the relevant statute, our view of FDA’s inconsistency with its past actions in this area and the retroactive effect of what we believe is a new policy at FDA as it relates to our situation. On May 28, 2015, the court granted our motion for summary judgment. The decision vacated the FDA’s denial of our claim for such exclusivity and remanded to the FDA for proceedings consistent with the decision. On July 22, 2015, Watson Laboratories Inc., the purported first Vascepa ANDA filer, sought to intervene and appeal the court’s decision. We and FDA opposed this intervention effort. The applicable courts denied Watson the relief sought and appeal periods have expired.
On May 31, 2016, in a reversal that FDA and we view as consistent with the court’s May 28, 2015 summary judgment motion, FDA determined that Vascepa is eligible for five-year, NCE marketing exclusivity. We believe this determination provides Vascepa with the benefits of NCE exclusivity afforded by statute. NCE exclusivity for Vascepa ran from its date of FDA approval on July 26, 2012 and extended until July 26, 2017. We believe the statutory NCE-related 30-month stay triggered by the 1-gram dose patent litigation following generic application submissions permitted on July 26, 2016 is scheduled to continue until January 26, 2020, seven-and-a-half years from FDA approval, unless such patent litigation is resolved against us sooner.
It is possible, but unlikely at this time given the time remaining in the 30-month stay, that FDA’s NCE determination and related 30-month stay could be challenged by interested parties. If challenged, we plan to vigorously defend exclusivity for Vascepa. Any such challenge could have a negative impact on our company and create uncertainty around the continued benefits associated with exclusivity that we believe are applicable to us under the Hatch-Waxman Amendments.
Regulatory exclusivity is in addition to exclusivity afforded by issued patents related to Vascepa.
Other Regulatory Matters
Manufacturing, sales, promotion, importation, and other activities related to approved products are also subject to regulation by numerous regulatory authorities, including, in the United States, the FDA, the Centers for Medicare & Medicaid Services, other divisions of the Department of Health and Human Services, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency, and state and local governments. Sales, marketing and scientific/educational programs must comply with the Food, Drug, and Cosmetic Act, the Anti-Kickback Statute, and the False Claims Act and similar state laws. Pricing and rebate programs must comply with the Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines or other penalties, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of product approvals, or refusal to allow a firm to enter into supply contracts, including government contracts. In addition, even if a firm complies with FDA and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA to modify or withdraw a product approval. Prohibitions or restrictions on sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
24
Changes in regulations or statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
Patents, Proprietary Technology, Trade Secrets
Our success depends in part on our ability to obtain and maintain intellectual property protection for our drug candidates, technology and know-how, and to operate without infringing the proprietary rights of others. Our ability to successfully implement our business plan and to protect our products with our intellectual property will depend in large part on our ability to:
|
|
• |
obtain, defend and maintain patent protection and market exclusivity for our current and future products; |
|
|
• |
preserve any trade secrets relating to our current and future products; |
|
|
• |
acquire patented or patentable products and technologies; and |
|
|
• |
operate without infringing the proprietary rights of third parties. |
We have prosecuted, and are currently prosecuting, multiple patent applications to protect the intellectual property developed during the Vascepa development program. As of the date of this report, we had 79 patent applications in the United States that have been either issued or allowed and more than 30 additional patent applications are pending in the United States. Such 79 allowed and issued applications include the following:
|
|
• |
2 issued U.S. patents directed to a pharmaceutical composition of Vascepa in a capsule that have terms that expire in 2020 and 2030, respectively; |
|
|
• |
1 issued U.S. patent covering a composition containing highly pure EPA that expires in 2021; |
|
|
• |
43 U.S. patents covering or related to the use of Vascepa in either the MARINE or ANCHOR populations that have terms that expire in 2030 or later; |
|
|
• |
11 U.S. patents covering or related to the use of Vascepa in the REDUCE-IT population with terms expiring in 2033 or later; |
|
|
• |
4 additional patents related to the use of a pharmaceutical composition comprised of free fatty acids to treat the ANCHOR patient population with a term that expires in 2030 or later; |
|
|
• |
2 additional patents related to the use of a pharmaceutical composition comprised of free fatty acids to treat the MARINE patient population with a term that expires in 2030; |
|
|
• |
3 additional patents related to the use of a pharmaceutical composition comprised of free fatty acids to treat the REDUCE-IT population expiring 2033; |
|
|
• |
1 additional patent related to a pharmaceutical composition comprised of free fatty acids and uses thereof to treat both the MARINE and ANCHOR patient populations with a term that expires in 2030; |
|
|
• |
1 additional patent related to the use of a pharmaceutical composition comprised of re-esterified EPA triglyceride to treat the REDUCE-IT population expiring 2033; |
|
|
• |
3 additional patents related to a formulation of EPA/DHA and uses thereof with a term that expires in 2030; |
|
|
• |
2 additional patents related to the use of Vascepa to treat obesity with a term that expires in 2034; |
|
|
• |
2 additional patents covering a pharmaceutical composition comprised of EPA and a hydroxyl compound with a term that expires in 2034; and |
|
|
• |
4 additional patents covering a new combination therapy comprised of EPA and another drug. |
A Notice of Allowance is issued after the USPTO makes a determination that a patent can be granted from an application. A Notice of Allowance does not afford patent protection until the underlying patent is issued by the USPTO. No assurance can be given that applications with issued notices of allowance will be issued as patents or that any of our pending patent applications will issue as patents. No assurance can be given that, if and when issued, our patents will prevent competitors from competing with Vascepa. For example, we may choose to not assert all issued patents in patent litigation and patents or claims within patents may be determined to be invalid.
We are the owner of the above-listed patents. We are also the exclusive licensee of certain patents owned by others covering products in development. To secure our debt under our outstanding royalty-like instrument, we have granted the holders of such instrument a security interest in our Vascepa-related patents.
25
We are also pursuing patent applications related to Vascepa in multiple jurisdictions outside the United States. We may be dependent in some cases upon third-party licensors to pursue filing, prosecution and maintenance of patent rights or applications owned or controlled by those parties, including, for example, under our collaboration with Mochida. It is possible that third parties will obtain patents or other proprietary rights that might be necessary or useful to us. In cases where third parties are first to invent a particular product or technology, or first to file after various provisions of the America Invents Act of 2011 went into effect on March 16, 2013, it is possible that those parties will obtain patents that will be sufficiently broad so as to prevent us from utilizing such technology or commercializing our current and future products.
Although we intend to make reasonable efforts to protect our current and future intellectual property rights and to ensure that any proprietary technology we acquire or develop does not infringe the rights of other parties, we may not be able to ascertain the existence of all potentially conflicting claims. Therefore, there is a risk that third parties may make claims of infringement against our current or future products or technologies. In addition, third parties may be able to obtain patents that prevent the sale of our current or future products or require us to obtain a license and pay significant fees or royalties in order to continue selling such products.
We may in the future discover the existence of products that infringe patents that we own or that have been licensed to us. If we were to initiate legal proceedings against a third party to stop such an infringement, such proceedings could be costly and time consuming, regardless of the outcome. No assurances can be given that we would prevail, and it is possible that, during such a proceeding, our patent rights could be held to be invalid, unenforceable or both. Although we intend to protect our trade secrets and proprietary know-how through confidentiality agreements with our manufacturers, employees and consultants, we may not be able to prevent parties subject to such confidentiality agreements from breaching these agreements or third parties from independently developing or learning of our trade secrets.
We anticipate that competitors may from time to time oppose our efforts to obtain patent protection for new technologies or to submit patented technologies for regulatory approvals. Competitors may seek to oppose our patent applications to delay the approval process or to challenge our granted patents, for example, by requesting a reexamination of our patent at the USPTO, or by filing an opposition in a foreign patent office, even if the opposition or challenge has little or no merit. For example, one of our patents was revoked in an opposition proceeding in Europe due to a determination of improper claim amendments under a provision of law not applicable in the United States. Such proceedings are generally highly technical, expensive, and time consuming, and there can be no assurance that such a challenge would not result in the narrowing or complete revocation of any patent of ours that was so challenged.
Employees
At February 22, 2019, we had 530 full-time employees employed in sales, marketing, general and administrative and research and development functions. We believe our relations with our employees are good.
Organizational Structure
At February 22, 2019, we had the following subsidiaries:
|
|
Subsidiary Name |
|
Country of Incorporation or Registration |
|
Proportion of Ownership Interest and Voting Power Held |
|
|
|
Amarin Pharmaceuticals Ireland Limited |
|
|
Ireland |
|
100% |
|
|
Amarin Pharma Inc. |
|
|
United States |
|
100% |
|
|
Amarin Neuroscience Limited |
|
|
Scotland |
|
100% |
|
|
Corsicanto II DAC |
|
|
Ireland |
|
100% |
|
|
Ester Neurosciences Limited |
|
|
Israel |
|
100% |
As of the date of this Annual Report on Form 10-K, our principal operating activities were being conducted by Amarin Corporation plc, together with Amarin Pharmaceuticals Ireland Limited and Amarin Pharma, Inc., with little to no operating activity being conducted by Amarin Neuroscience Limited, Corsicanto II DAC, or Ester Neurosciences Limited. Corsicanto DAC (formerly Corsicanto Limited) was liquidated in January 2019 pursuant to a resolution of Amarin Corporation plc as a sole shareholder.
Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to reports filed pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended, are made available free of charge on or through our website at www.amarincorp.com as soon as reasonably practicable after such reports are filed with, or furnished to, the Securities and Exchange Commission, or SEC. The SEC also maintains a website, www.sec.gov, that contains reports and other
26
information regarding issuers that file electronically with the SEC. We are not, however, including the information contained on our website, or information that may be accessed through links on our website, as part of, or incorporating such information by reference into, this Annual Report on Form 10-K.
Financial Information
The financial information required under this Item 1 is incorporated herein by reference to Item 8 of this Annual Report on Form 10-K.
This Annual Report on Form 10-K contains forward-looking information based on our current expectations. Because our actual results may differ materially from any forward-looking statements that we make or that are made on our behalf, this section includes a discussion of important factors that could affect our actual future results, including, but not limited to, our ability to successfully commercialize Vascepa, our capital resources, the progress and timing of our clinical programs, the safety and efficacy of our product candidates, risks associated with regulatory filings, the potential clinical benefits and market potential of our product candidates, commercial market estimates, future development efforts, patent protection, effects of healthcare reform, reliance on third parties, effects of tax reform, and other risks set forth below.
Risks Related to the Commercialization and Development of Vascepa
We are substantially dependent upon sales of Vascepa in the United States.
As a result of our reliance on a single product, Vascepa® (icosapent ethyl) capsules, and our primary focus on the U.S. market in the near-term, much of our near-term results and value as a company depends on our ability to execute our commercial strategy for Vascepa in the United States. If commercialization efforts for Vascepa do not meet expectations, our business could be materially and adversely affected.
Even if we are able to successfully develop Vascepa outside the United States or develop additional products from our research and development efforts, the development time cycle for products typically takes several years. If we seek to diversify our development programs or product offerings through licensing or acquisitions, such transactions are also time-consuming, dilutive to existing shareholdings, and can be disruptive to operations. These dynamics can restrict our ability to respond rapidly to adverse business conditions for Vascepa. If demand for Vascepa does not meet expectations and we are not successful with development, we may not have the ability to effectively shift our resources to the development of alternative products or do so in a timely manner without suffering material adverse effects on our business. As a result, the lack of alternative markets and products we develop could constrain our ability to generate revenues and achieve profitability.
Factors out of our control make it more difficult for Vascepa to achieve a level of market acceptance by physicians, patients, healthcare payors and others in the medical community necessary for commercial success.
In January 2013, we launched Vascepa based on the U.S. Food and Drug Administration, or FDA, approval of our MARINE indication, for use as an adjunct to diet to reduce triglyceride levels in adult patients with severe (TG ≥500 mg/dL) hypertriglyceridemia. Guidelines for the management of very high triglyceride levels suggest that the primary goal of reducing triglyceride levels in this patient population is reduction in the risk of acute pancreatitis. A secondary goal for this patient population is to reduce cardiovascular risk. The effect of Vascepa on cardiovascular mortality and morbidity, or the risk for pancreatitis, in patients with severe hypertriglyceridemia has not been determined and our FDA-approved labeling and promotional efforts state these facts.
In August 2015, based on a federal court order, we began communicating promotional information beyond the MARINE indication to healthcare professionals in the United States for the treatment of patients with high (TG ≥200 mg/dL and <500 mg/dL) triglyceride levels who are also on statin therapy for elevated low-density lipoprotein cholesterol, or LDL-C, levels, based on results from the ANCHOR study of Vascepa. Many patients with high triglycerides also have other lipid level abnormalities such as high cholesterol and are on statin therapy. FDA did not approve Vascepa for use in this population due to the uncertain effect of pharmaceutically induced triglyceride reduction in this patient population on cardiovascular risk reduction, the ultimate targeted clinical benefit. Our promotional efforts disclose this fact and what we view as truthful and non-misleading information on the current state of research on both triglyceride reduction and the active pharmaceutical ingredient, or API, in Vascepa, EPA, as each relate to the potential of Vascepa to reduce cardiovascular risk.
In September 2018, we announced topline results from the REDUCE-IT™ (Reduction of Cardiovascular Events with EPA—Intervention Trial) cardiovascular (CV) outcomes study of Vascepa. In November 2018, we announced the primary results of our REDUCE-IT cardiovascular outcomes study confirming 25% relative risk reduction for the topline primary endpoint result with multiple robust demonstrations of efficacy, including 20% reduction in cardiovascular death. REDUCE-IT was a multinational, prospective,
27
randomized, double-blind, placebo-controlled study, enrollment for which started in November 2011. REDUCE-IT investigated the effects of Vascepa on CV risk in statin-treated adults with well-controlled LDL-C 41-100 mg/dL (median baseline LDL-C: 75 mg/dL) and other CV risk factors, including persistent elevated TG 150-499 mg/dL (median baseline TG: 216 mg/dL). REDUCE-IT topline results showed the trial met its primary endpoint demonstrating an approximately 25% relative risk reduction, to a high degree of statistical significance (p<0.001), in major adverse cardiovascular events (MACE) in the intent-to-treat patient population with use of Vascepa 4 grams/day as compared to placebo. MACE events were defined as a composite of cardiovascular death, nonfatal myocardial infarction (MI), nonfatal stroke, coronary revascularization, or unstable angina requiring hospitalization. This result was supported by robust demonstrations of efficacy across multiple secondary endpoints. Vascepa was well tolerated in REDUCE-IT with a safety profile generally consistent with clinical experience associated with omega-3 fatty acids and current FDA-approved labeling. The proportions of patients experiencing adverse events and serious adverse events in REDUCE-IT were similar between the active and placebo treatment groups. Based on the final positive results of REDUCE-IT, we plan to seek additional indicated uses for Vascepa in the United States and to continue to develop Vascepa commercially in major markets around the world.
Even though we have released positive results from the REDUCE-IT trial, our approved label for Vascepa in the United States currently remains unchanged pending additional interactions and review by the FDA. A failure to obtain an expanded label may make it more difficult for Vascepa to gain market acceptance by physicians, patients, healthcare payors and others in the medical community. If Vascepa does not achieve an adequate level of acceptance, we may not generate product revenues sufficient to become profitable. The degree of market acceptance of Vascepa for the MARINE indication and in ANCHOR patients and in any future indications and uses based on the REDUCE-IT trial or otherwise will depend on a number of factors, including:
|
|
• |
the perceived efficacy and safety of Vascepa by prescribing healthcare professionals, as compared to no treatment and as compared to alternative treatments in various at-risk patient populations, both as studied in clinical trials of Vascepa such as MARINE, ANCHOR and REDUCE-IT and not studied but for which the benefit/risk profile may be viewed as positive; |
|
|
• |
peer review of REDUCE-IT results and publication of results in one or more medical journals over time; |
|
|
• |
the FDA’s review and analysis of the results of REDUCE-IT; |
|
|
• |
our ability to offer Vascepa for sale at competitive prices; |
|
|
• |
convenience and ease of administration compared to alternative treatments; |
|
|
• |
the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
|
|
• |
the scope, effectiveness and strength of product education, marketing and distribution support, including our sales and marketing team; |
|
|
• |
publicity concerning Vascepa or competing products; |
|
|
• |
our ability to continually promote Vascepa in the United States outside of FDA-approved labeling and the related perception thereof; |
|
|
• |
sufficient third-party coverage or reimbursement for on-label use, and for permitted off-label use, the third-party coverage or reimbursement which was not addressed in the scope of the August 2015 court declaration or related settlement; |
|
|
• |
natural disasters that can inhibit our ability to promote Vascepa regionally and can negatively affect product demand by creating obstacles for patients to seek treatment and fill prescriptions; |
|
|
• |
new policies or laws affecting Vascepa sales, such as state and federal efforts to affect drug pricing and provide or remove healthcare coverage that includes reimbursement for prescription drugs; and |
|
|
• |
the actual efficacy of the product and the prevalence and severity of any side effects, including any limitations or warnings contained in Vascepa’s approved labeling. |
As with any cardiovascular outcomes trial, over time further REDUCE-IT data assessment and data release will yield additional useful information to inform greater understanding of study outcome. That additional data may exceed, meet or not meet investor expectations. If the additional data do not meet expectations, the perception of REDUCE-IT results and Vascepa may suffer and our stock price may decline.
In September 2018, we announced topline results from the REDUCE-IT trial showing that the trial met its primary endpoint demonstrating an approximately 25% relative risk reduction, to a high degree of statistical significance (p<0.001), in MACE in the intent-to-treat patient population with use of Vascepa 4 grams/day as compared to placebo. More detailed presentation of REDUCE-IT results was first made at the 2018 Scientific Sessions of the American Heart Association (AHA) on November 10, 2018 with such results concurrently published in The New England Journal of Medicine. Additional data assessment and data release will yield additional useful information to inform greater understanding of study outcome. Generally, trial data assessment sufficient to convey a complete picture of trial data typically takes several months and can take years to complete and publish. When new data are assessed and released it could exceed, match or may not meet investor expectations. For example, a presentation of additional analyses of
28
REDUCE-IT results is scheduled for the American College of Cardiology (ACC) 2019 Scientific Sessions on March 18, 2019 in New Orleans, Louisiana. This presentation and additional data may exceed, match or may not meet investor expectations.
In addition, the same set of data can sometime be interpreted to reach conclusions that conflict with our conclusions, as was the case when FDA reviewed earlier cardiovascular outcomes trials of other drugs in the context of the effects of triglyceride lowering agents on cardiovascular risk reduction.
Aspects that could change and impact the final evaluation of the totality of the efficacy and safety data from REDUCE-IT may include some or all of the following:
|
|
• |
the magnitude of the treatment benefit on the primary composite endpoint, its components, secondary endpoints and the primary and secondary risk prevention cohorts; |
|
|
• |
consideration of which components of the composite or secondary endpoints have the most clinical significance; |
|
|
• |
the consistency of the primary and secondary outcomes; |
|
|
• |
the consistency of findings across cohorts and important subgroups; |
|
|
• |
safety considerations and risk/benefit considerations; |
|
|
• |
consideration of REDUCE-IT results in the context of other clinical studies; |
|
|
• |
consideration of the cumulative effect of Vascepa in studied patients; and |
|
|
• |
study conduct and data quality, integrity and consistency. |
If release of additional data do not meet expectations, the perception of REDUCE-IT results and the perceived value of Vascepa may suffer. If this occurs our business could suffer and our stock price could significantly decline.
Clinical trials that we or potential partners conduct, including the REDUCE-IT trial, may not provide sufficient safety and efficacy data to obtain the requisite regulatory approvals for product candidates or to achieve a level of market acceptance by physicians, patients, healthcare payors and others in the medical community necessary for commercial success if we obtain regulatory approval.
On November 10, 2018, we announced the primary results of our REDUCE-IT cardiovascular outcomes study confirming 25% relative risk reduction for the topline primary endpoint result with multiple robust demonstrations of efficacy, including 20% reduction in cardiovascular death. Major cardiovascular outcomes studies like REDUCE-IT typically motivate the medical community to search for ways to fit the results into the mosaic of prior studies considered as successful, like JELIS, and CANTOS, and their associated mechanisms of action, and to also distinguish results from prior failed studies, like what the authors of The New England Journal of Medicine editorial on REDUCE-IT referred to as the “parade of failed cardiovascular outcome trials of fish oils.” It is important for the medical community to understand related science on the unique active pharmaceutical ingredient in Vascepa, icosapent ethyl, and REDUCE-IT. As with any clinical study, as the medical community and regulators such as the FDA review and analyze REDUCE-IT study data, dialogue is expected to continue with respect to the reliability of REDUCE-IT data and the study quality that could adversely affect our product development, regulatory review, market or medical community acceptance, and level of payor reimbursement in the event of an expansion of the Vascepa label. Likewise, public perception of the REDUCE-IT results and Vascepa may be affected.
For example, in the REDUCE-IT trial, cardiovascular benefits appeared not to be influenced significantly by TG levels at baseline (above or below 150 mg/dL baseline range) or as achieved at one year, potentially suggesting mechanisms at work with use of Vascepa that are independent of baseline TG levels or therapy-driven reduction in TG levels. Determination of the mechanisms responsible for the benefit shown in REDUCE-IT was not the focus or purpose of the study. As summarized from the primary results of REDUCE-IT in The New England Journal of Medicine, potential Vascepa mechanisms of action at work in REDUCE-IT may include TG reduction, anti-thrombotic effects, antiplatelet or anticoagulant effects, membrane-stabilizing effects, effects on stabilization and/or regression of coronary plaque and inflammation reduction, each as supported by earlier stage mechanistic studies.
In addition, the median change in LDL cholesterol level from baseline was 6.6% (5.0 mg/dL; p < 0.001) on a placebo-corrected basis reflecting an increase of 3.1% (+2.0 mg/dL) in the Vascepa group and an increase of 10.2% (+7.0 mg/dL) in the placebo group. Increases in the placebo group relative to the Vascepa group were also observed in other parameters classically measured in such studies but with uncertain relevance to cardiovascular outcomes. An upward drift in LDL cholesterol and such other parameter has been commonly, although not always, observed in statin-stabilized patients across numerous studies within varying patient populations, and many have estimated LDL cholesterol increases of at least 6% and ranging up to more than 30%. Factors
29
cited as potentially contributing to this circumstance include decreased drug and lifestyle regimen compliance, physiological compensation for drug-induced lipid changes, regression to the mean, intraindividual variability, lab variability, genetics, metabolic state, disease state, age, and season. If light liquid paraffin oil, or mineral oil, used as the placebo in REDUCE-IT adversely affected statin absorption or other parameters in some patients as is asserted by certain critics of the study, this could be theorized to have contributed to differences in outcomes between the groups and leave open the possibility that the placebo used in the trial was not biologically inert. These and other observations, whether scientifically reliable or not, may negatively impact how these trial results are interpreted by regulators, the medical community and third-party payors. This is the case notwithstanding that a post hoc analysis of REDUCE-IT data published in The New England Journal of Medicine showed no material differences in the primary and key secondary endpoint event rates for placebo patients with an increase in LDL-C at one year versus those with no change or a decrease, and also suggested a similar relative risk reduction regardless of whether there was an increase in LDL cholesterol level among the patients in the placebo group. Data generated by Amarin after, but supporting, this analysis reflect that patient-by-patient differences in LDL cholesterol levels from baseline to Year 1 included some patients with increases, some patients with decreases and others with no change in both the Vascepa arm and the placebo arm of the REDUCE-IT study. If mineral oil affected statin absorption significantly, it is reasonable to expect that such effect might be evident in all patients on placebo (i.e., if mineral oil had a definitive effect one would expect LDL cholesterol increases would be consistently observed among patients in the placebo arm) rather than the observed mixed results that include many patients with LDL cholesterol decreases or lack of change in LDL cholesterol. Moreover, as the authors of the primary results publication on REDUCE-IT in The New England Journal of Medicine noted, the relatively small differences in LDL cholesterol levels between the groups would not be likely to explain the 25% lower MACE risk observed with Vascepa and JELIS, an over 18,000 patient cardiovascular outcomes study in Japan of a highly-pure EPA product similar to Vascepa, that, previously demonstrated a 19% risk reduction without a mineral oil placebo.
Consistent with our SPA for REDUCE-IT agreed to with the FDA, the trial subjects in the placebo arm of REDUCE-IT were given light liquid paraffin oil, or mineral oil, to mimic the color and consistency of Vascepa. We also used mineral oil in the placebo arms of our MARINE and ANCHOR trials. During the public advisory committee meeting held by FDA as part of its review of our ANCHOR sNDA, a discussion regarding observed, nominally statistically significant changes from baseline in an adverse direction, while on background statin therapy, in certain lipid parameters, including triglycerides, in the placebo group, led to further discussion about the possibility that the mineral oil placebo used in the ANCHOR trial (and in the REDUCE-IT trial) might not be biologically inert and might be viewed as artificially exaggerating the clinical effect of Vascepa when measured against placebo in the ANCHOR trial. Ultimately, no strong evidence for biological activity of mineral oil was identified by the FDA before its approval of Vascepa after review of the MARINE and ANCHOR trials and consideration of other data regarding mineral oil. The FDA, early in the course of the REDUCE-IT trial, directed the independent data monitoring committee, or DMC, for REDUCE-IT to periodically review unblinded lipid data to monitor for signals that the placebo might not be inert. After each such quarterly unblinded safety analysis and review meeting to date, the DMC recommended to continue the REDUCE-IT study as planned. Each of these DMC recommendations has been shared with FDA. In addition, following discussions on this topic in October 2013 in connection with the FDA’s review of our supplemental new drug application for our ANCHOR study, the FDA did not seek to require that we include any qualification related to the use of mineral oil as a placebo in REDUCE-IT at the time of our March 2016 amendment to the REDUCE-IT SPA. As noted, importantly and consistently, JELIS, an over 18,000 patient cardiovascular outcomes study in Japan of a highly-pure EPA product similar to Vascepa previously demonstrated a 19% risk reduction without a mineral oil placebo.
As with any cardiovascular outcomes trial, further REDUCE-IT data assessment and data release could yield additional useful information to inform a greater understanding of the trial outcome. Further detailed data assessment by Amarin and regulatory authorities will continue and take several months or more to complete and record. The final evaluation of the totality of the efficacy and safety data from REDUCE-IT may include some or all of the following, as well as other considerations: new information affecting the degree of treatment benefit on studied endpoints; study conduct and data robustness, quality, integrity and consistency; additional safety data considerations and risk/benefit considerations; consideration of the cumulative effect of Vascepa in studied patients; and consideration of REDUCE-IT results in the context of other clinical studies. That additional data may exceed, meet or not meet the expectations of regulators, the medical community and third-party payors.
If Vascepa’s specific mechanism of action shown in the REDUCE-IT study or the potential effects of the mineral oil used in the placebo arm of REDUCE-IT remains uncertain, or any additional data from the REDUCE-IT study do not meet expectations, the perception of REDUCE-IT results and Vascepa may suffer and could adversely affect our product development, regulatory review, market or medical community acceptance, level of payor reimbursement in the event of an expansion of the Vascepa label, or the public perception of the REDUCE-IT results and Vascepa, any of which could have a material adverse effect on our business and financial condition and our stock price may decline.
Our current and planned commercialization efforts in the United States may not be successful in increasing sales of Vascepa.
Prior to REDUCE-IT topline results announcement in September 2018, our sales team consisted of approximately 170 sales professionals, including sales representatives and their managers. We have recently increased the size of our sales force to approximately 440 sales professionals, including approximately 400 sales representatives, pursuant to positive REDUCE-IT results
30
and are expanding our promotion of Vascepa. This sales team promotes Vascepa to a limited group of physicians and other healthcare professionals in select geographies in the United States. Even after planned expansion, this sales team is not large enough to call upon all physicians. We intend to further expand the promotion of Vascepa following our assumed label expansion for Vascepa, subject to FDA review and approval of our sNDA.
In May 2014, we began co-promoting Vascepa in the United States with Kowa Pharmaceuticals America, Inc. under a co-promotion agreement we entered into in March 2014, which we amended in July 2017. Under the agreement, Kowa Pharmaceuticals America, Inc. co-promoted Vascepa in conjunction with its promotion of its primary product, a branded statin for patients with high cholesterol, along with our sales professionals based on a plan designed to focus on select sales territories that we believed had demonstrated the greatest potential for Vascepa sales growth, increasing both the number of sales targets reached and the frequency of sales calls on existing sales targets. However, the commercialization of pharmaceutical products is a complex undertaking, and we had very limited experience as a company operating in this area and co-promoting a pharmaceutical product with a partner.
Furthermore, our agreement with Kowa Pharmaceuticals America, Inc. was designed such that Kowa’s co-promotion of Vascepa ceased at the end of 2018. The parties mutually agreed not to renew the agreement. If our newly expanded sales team are not at least equally capable, our sales may be negatively impacted.
In addition to sales force expansion in the United States, Amarin plans to work with its international partners to support regulatory efforts outside the United States based on REDUCE-IT results. We will again need to overcome challenges associated with rapidly hiring and training personnel and managing larger teams of people.
Factors related to building and managing a sales and marketing organization that can inhibit our efforts to successfully commercialize Vascepa include:
|
|
• |
our inability to attract and retain adequate numbers of effective sales and marketing personnel; |
|
|
• |
our inability to adequately train our sales and marketing personnel, in particular as it relates to various healthcare regulatory requirements applicable to the marketing and sale of pharmaceutical products and the court declaration that we believe enables us to expand marketing efforts for Vascepa, and our inability to adequately monitor compliance with these requirements; |
|
|
• |
the inability of our new sales personnel, working for us as a new market entrant, to obtain access to or persuade adequate numbers of physicians to prescribe Vascepa; |
|
|
• |
the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; |
|
|
• |
an inability by us or our partners to obtain regulatory and marketing approval or establish marketing channels in foreign jurisdictions; and |
|
|
• |
unforeseen costs and expenses associated with operating a new independent sales and marketing organization. |
If we are not successful in our efforts to market and sell Vascepa, our anticipated revenues will be materially and negatively affected, and we may not obtain profitability, may need to cut back on research and development activities or need to raise additional funding that could result in substantial dilution.
Our past and future off-label promotion of Vascepa could subject us to additional regulatory scrutiny and present unforeseen risks.
The Federal Food, Drug, and Cosmetic Act, or FDCA, has been interpreted by the FDA and the U.S. government to make it illegal for pharmaceutical companies to promote their FDA approved products for uses that have not been approved by the FDA. Companies that market drugs for off-label uses or indications have been subject to related costly litigation, criminal penalties and civil liability under the FDCA and the False Claims Act. However, recent case law has called into question the extent to which government in the United States, including FDA, can, and is willing to seek to, prevent truthful and non-misleading speech related to off-label uses of FDA-approved products such as Vascepa.
In May 2015, we and a group of independent physicians filed a lawsuit against the FDA seeking a federal court declaration that would permit us and our agents to promote to healthcare professionals the use of Vascepa in the ANCHOR population and promote on the potential of Vascepa to reduce the risk of cardiovascular disease so long as the promotion is truthful and non-misleading. This use of Vascepa at issue reflected recognized medical practice but was not approved by the FDA and is thus not covered by current FDA-approved labeling for the drug. Promotion of an off-label use has generally been considered by the FDA to be illegal under the FDCA. The lawsuit, captioned Amarin Pharma, Inc., et al. v. Food & Drug Administration, et al., 119 F. Supp. 3d 196 (S.D.N.Y. 2015), was filed in the United States District Court for the Southern District of New York. In the lawsuit, we contended principally that FDA regulations limiting off-label promotion of truthful and non-misleading information are unconstitutional under the freedom of speech
31
clause of the First Amendment to the U.S. Constitution as applied in the case of our proposed promotion of Vascepa. The physicians in the suit regularly treated patients at risk of cardiovascular disease and, as the complaint contended, have First Amendment rights to receive truthful and non-misleading information from Amarin. The suit was based on the principle that better informed physicians make better treatment decisions for their patients. The FDA opposed this lawsuit but did not dispute the veracity of the subject ANCHOR clinical trial data (the safety data from which was already and currently is in FDA-approved labeling of Vascepa) or the peer-reviewed research related to Vascepa and the potential for cardiovascular risk reduction.
In August 2015, we were granted preliminary relief in this lawsuit through the court’s declaratory judgment that confirmed we may engage in truthful and non-misleading speech promoting the off-label use of Vascepa to healthcare professionals, i.e., to treat patients with persistently high triglycerides, and that such speech may not form the basis of a misbranding action under the FDCA. In August 2015, we began to communicate promotional information beyond the MARINE indication to healthcare professionals in the United States as permitted by this court declaration. The FDA did not appeal the court’s ruling.
In March 2016, we settled this litigation under terms by which the FDA and the U.S. government agreed to be bound by the conclusions from the federal court order that we may engage in truthful and non-misleading speech promoting the off-label use of Vascepa and that certain statements and disclosures that we proposed to make to healthcare professionals were truthful and non-misleading. As part of the settlement, given, as expressed in the court’s opinion, that the dynamic nature of science and medicine is that knowledge is ever-advancing and that a statement that is fair and balanced one day may become incomplete or otherwise misleading in the future as new studies are done and new data is acquired, we agreed that we bear the responsibility to ensure that our communications regarding off-label use of Vascepa remain truthful and non-misleading, consistent with the federal court ruling.
While we believe we are now permitted under applicable law to more broadly promote Vascepa, the FDA-approved labeling for Vascepa did not change as a result of this litigation and settlement, and neither government nor other third-party coverage or reimbursement to pay for the off-label use of Vascepa promoted under the court declaration was required. Based on our communications with the FDA, we expect that the FDA’s review and analysis of our final positive results from the REDUCE-IT outcomes study will be required for FDA-approved label expansion for Vascepa. However, we proactively communicate results from the REDUCE-IT trial in a manner we believe is truthful and non-misleading and thus protected under the freedom of speech clause of the First Amendment to the United States Constitution.
Even though we have the benefit of a final settlement in this litigation, our promotion is still subject to a high, perhaps abnormally high, degree of scrutiny to ensure that our promotion remains within the scope covered by the settlement. For example, under the settlement, we remain responsible for ensuring our speech is truthful and non-misleading. Data arising from studies of drug products are complex, such as the many studies that we believe show supportive but not conclusive research on the potential connection between the effects of EPA, the active ingredient in Vascepa, and cardiovascular risk reduction (e.g., the JELIS trial of a highly-pure EPA product in Japan by Mochida Pharmaceutical Co., Ltd., or Mochida, and other data using a variety of levels of evidence that connect EPA to favorable effects toward reduced cardiovascular risk). We, the FDA, the U.S. government, our competitors and other interested parties may not agree on the truthfulness and non-misleading nature of our promotional materials with respect to the outcome of these trials or other direct or indirect claims we make about Vascepa. Likewise, the FDA, the U.S. government, our competitors and other interested parties may not agree on the truthfulness and non-misleading nature of our promotional materials related to the REDUCE-IT results. Federal and state governments or agencies may also seek to find other means to prevent our promotion of unapproved truthful and non-misleading information about Vascepa. If our promotional activities or other operations are found to be in violation of any law or governmental regulation through existing or new interpretations, we may be subject to prolonged litigation, penalties, including civil and criminal penalties, damages, fines and the curtailment or restructuring of our operations. Also, if governmental parties or our competitors view our claims as misleading or false, we could also be subject to liability based on fair competition-based statutes, such as the Lanham Act. Any of such negative circumstances could adversely affect our ability to operate our business and our results of operations.
We may not be able to compete effectively against our competitors’ pharmaceutical products.
The biotechnology and pharmaceutical industries are highly competitive. There are many pharmaceutical companies, biotechnology companies, public and private universities and research organizations actively engaged in the research and development of products that may be similar to our products. It is probable that the number of companies seeking to develop products and therapies similar to our products will increase. Many of these and other existing or potential competitors have substantially greater financial, technical and human resources than we do and may be better equipped to develop, manufacture and market products. These companies may develop and introduce products and processes competitive with or superior to ours. In addition, other technologies or products may be developed that have an entirely different approach or means of accomplishing the intended purposes of our products, which might render our technology and products noncompetitive or obsolete.
Our competitors both in the United States and abroad include large, well-established pharmaceutical and generic companies, specialty and generic pharmaceutical sales and marketing companies, and specialized cardiovascular treatment companies. GlaxoSmithKline plc currently sells Lovaza®, a prescription-only omega-3 fatty acid indicated for patients with severe hypertriglyceridemia, which was approved by FDA in 2004 and has been on the market in the United States since 2005. Multiple
32
generic versions of Lovaza are available in the United States. Other large companies with competitive products include AbbVie, Inc., which currently sells Tricor® and Trilipix® for the treatment of severe hypertriglyceridemia and Niaspan®, which is primarily used to raise high-density lipoprotein cholesterol, or HDL-C, but is also used to lower triglycerides. Multiple generic versions of Tricor, Trilipix and Niaspan are also available in the United States. We compete with these drugs, and in particular, multiple low-cost generic versions of these drugs, in our FDA-approved indicated use and in off-label uses, such as to beneficially affect lipid levels in patients with persistent high triglyceride levels after statin therapy with the aim of potentially lowering cardiovascular risk beyond statin therapy.
In addition, in May 2014, Epanova® (omega-3-carboxylic acids) capsules, a free fatty acid form of omega-3 (comprised of 55% EPA and 20% DHA), was approved by the FDA for patients with severe hypertriglyceridemia. Epanova was developed by Omthera Pharmaceuticals, Inc., and is now owned by AstraZeneca Pharmaceuticals LP (AstraZeneca). Also, in April 2014, Omtryg, another omega-3-acid fatty acid composition developed by Trygg Pharma AS, received FDA approval for severe hypertriglyceridemia. Neither Epanova nor Omtryg have been commercially launched, but could launch at any time. Each of these competitors, other than potentially Trygg, has greater resources than we do, including financial, product development, marketing, personnel and other resources. Neither Epanova or Omtryg have been commercially launched.
AstraZeneca is currently conducting a long-term outcomes study to assess Statin Residual Risk Reduction With EpaNova in HiGh Cardiovascular Risk PatienTs With Hypertriglyceridemia (STRENGTH). The study is a randomized, double-blind, placebo-controlled (corn oil), parallel group design that is believed to have enrolled approximately 13,000 patients with hypertriglyceridemia and low HDL and high risk for cardiovascular disease randomized 1:1 to either corn oil plus statin or Epanova plus statin, once daily, for approximately 3-5 years as determined when the number of major adverse cardiovascular event outcomes is reached. The STRENGTH study is estimated to be completed in 2020, but it could be stopped earlier if, for example, it generates an overwhelming efficacy result. In addition, in March 2017, Kowa Research Institute (a subsidiary of the Japanese company Kowa Co., Ltd) initiated a phase III cardiovascular outcomes trial titled PROMINENT examining the effect of pemafibrate (experimental name K-877) in reducing cardiovascular events in Type II diabetic patients with hypertriglyceridemia. Kowa Research Institute has publicly estimated study completion in May 2022, and if successful, U.S. regulatory approval is estimated in mid-2023.
During 2018, two outcomes studies were completed of omega-3 mixtures which both failed to achieve their primary endpoints of cardiovascular risk reduction and two meta-analyses were published showing that omega-3 mixtures are not effective in lowering cardiovascular risk. Results of these failed outcomes studies and analysis, while not done with Vascepa, may negatively affect sales of Vascepa. For example, results of VITamin D and OmegA-3 TriaL (VITAL), as announced immediately before the presentation of REDUCE-IT results at the 2018 Scientific Sessions of the American Heart Association (AHA) on November 10, 2018, failed to achieve its primary endpoint of lowering cardiovascular events. VITAL was an NIH funded randomized double-blind, placebo-controlled, 2x2 factorial trial of 2000 IU per day of vitamin D3 and 1 gram per day of omega-3 fatty acid supplementation (Lovaza) for the primary prevention of cancer and cardiovascular disease in a nationwide USA cohort of 25,874 adults not selected for elevated cardiovascular or cancer risk.
Likewise, in 2018, results from A Study of Cardiovascular Events iN Diabetes (ASCEND) trial were released and showed negligible results for omega-3 fatty acids 1 gram daily. ASCEND was a British Heart Foundation funded 2x2 factorial design, randomized study to assess whether aspirin 100 mg daily versus placebo and separately, omega-3 fatty acids 1 gram daily versus placebo, reduce the risk of cardiovascular events in a nationwide UK cohort of over 15,000 individuals with diabetes who do not have atherosclerotic cardiovascular disease. In addition, VITAL showed that supplementation with either omega-3 fatty acid at a dose of 1 gram per day or vitamin D3 at a dose of 2000 IU per day was not effective for primary prevention of CV or cancer events among healthy middle-aged men and women across 5 years of follow up.
In meta-analysis, presented in 2018 by the Cochran Foundation and separately as published in JAMA, additional omega-3 studies were evaluated. Similar to the VITAL and ASCEND studies, most of the studies in these omega-3 meta-analyses were of omega-3 mixtures, including DHA, and most were studies of relatively low doses of omega-3 as is associated with dietary supplementation and/or they studied relatively low risk patient populations. The exception was the JELIS study, conducted in Japan, of highly pure EPA which demonstrated a positive outcome benefit. The negative results from such omega-3 mixture studies could create misleading impressions about the use of omega-3s generally, including Vascepa, despite REDUCE-IT positive results and the highly-pure and stable EPA active ingredient in Vascepa and its higher dose regimen.
We are also aware of other pharmaceutical companies that are developing products that, if successfully developed, approved and marketed, would compete with Vascepa. Acasti Pharma, or Acasti, a subsidiary of Neptune Technologies & Bioresources Inc., announced in December 2015 that it intends to pursue a regulatory pathway under section 505(b)(2) of the FDCA for its omega-3 prescription drug candidate, CaPre® (omega-3 phospholipid), derived from krill oil, for the treatment of hypertriglyceridemia. In September 2016, Acasti announced positive results from its pivotal bioavailability bridging study comparing CaPre to Lovaza, establishing a scientific bridge between the two that is expected to support the feasibility of a 505(b)(2) regulatory pathway. Acasti initiated a Phase 3 clinical program (TRILOGY) to assess the safety and efficacy of CaPre in patients with very high (≥500 mg/dL)
33
triglycerides in the first quarter of 2018. Acasti completed enrollment in Q4 2018 and study completion is expected by the end of 2019. We believe Micelle BioPharma Inc., or Micelle, is also developing potential treatments for hypertriglyceridemia based on omega-3 fatty acids. To our knowledge, Micelle, after acquiring SC401 from Sancilio & Company, or Sancilio, is pursuing a regulatory pathway under section 505(b)(2) of the FDCA for its product and submitted an Investigational New Drug Application, or IND, in July 2015. Micelle (Sancilio) completed two pharmacokinetic studies and Phase 2 bioavailability studies (FASTR I&II), with one comparing SC401 to Lovaza. We expect the company or a potential partner to initiate a pivotal clinical Phase 3 study as the next step in development.
Matinas BioPharma, Inc. is developing an omega-3-based therapeutic (MAT9001) for the treatment of severe hypertriglyceridemia and mixed dyslipidemia. In the fourth quarter of 2014, Matinas BioPharma, Inc. filed an IND with the FDA to conduct a human study in the treatment of severe hypertriglyceridemia and, in June 2015, the company announced topline results for its head-to-head comparative pharmacokinetic and pharmacodynamic study of MAT9001 versus Vascepa in patients under conditions inconsistent with the FDA-approved label for Vascepa and presented results based on biomarker modification without outcomes data. In September 2017, Matinas announced that it will be seeking a partner company to develop and commercialize MAT9001. Akcea Therapeutics/Ionis Pharmaceuticals (formerly Isis Pharmaceuticals), or Akcea/Ionis, in August 2018 announced the receipt of a complete response letter from the FDA for WAYLIVRA™ (volanesorsen), a drug candidate administered through weekly subcutaneous injections, for the treatment of familial chylomicronemia syndrome (FCS). Akcea will continue to work with the FDA on the path forward for Waylivra for the treatment of FCS. Waylivra continues to be developed for the treatment of familial partial lipodystrophy (FPL).
A Phase 3 trial is currently ongoing studying Waylivra (volanesorsen) in patients with FPL (BROADEN trial). Akcea/Ionis expects to file an NDA for FPL in 2019. In January 2017, Akcea/Ionis announced a strategic collaboration and option agreement with Novartis whereby Novartis will help develop (including funding cardiovascular outcomes studies) and commercialize products emerging from this collaboration, including Waylivra (volanesorsen). In June 2018, Gemphire Therapeutics announced positive topline results from a Phase 2b trial (INDIGO-1) of its drug candidate, gemcabene, in patients with severe hypertriglyceridemia. Gemcabene is an oral, once-daily pill for a number of hypercholesterolemic populations and severe hypertriglyceridemia. Gemphire announced plans to initiate a Phase III study for homozygous familial hypercholesterolemia (HoFH), heterozygous familial hypercholesterolemia (HeFH) and non-familial hypercholesterolemia in ASCVD patients in the second half of 2018. In August 2018, the FDA requested that Gemphire conduct an additional long-term toxicity study before commencing any further clinical testing, thereby effectively placing gemcabene on clinical hold. Zydus Cadila has a Phase 2 development program for its lead molecule, Saroglitazar, in various indications, including severe hypertriglyceridemia in the United States. In August 2018, the company announced that it had suspended the Phase 2 trial in the severe hypertriglyceridemia indication due to study enrollment issues, while it continues development activities in other indications. The product is approved in India under the name Lipaglyn® for the treatment of hypertriglyceridemia and diabetic dyslipidemia. We are also aware that bezafibrate has been licensed by Intercept Pharmaceuticals to be further developed and potentially launched in the United States market.
Based on prior communications from the FDA, including communications in connection with its review of the ANCHOR indication for Vascepa, it is our understanding that the FDA is not prepared to approve any therapy for treatment of cardiovascular risk based on biomarker modification without outcomes study data, with the potential exception of therapies which lower LDL-cholesterol. In particular, it is our understanding that the FDA is not prepared to approve any therapy based on data demonstrating lowering of triglyceride levels. In our view, this position from the FDA is unlikely to change based on the REDUCE-IT study particularly in light of the independence of the positive benefit demonstrated in the REDUCE-IT study from triglyceride levels and benefit from the REDUCE-IT study supporting that the positive effects of Vascepa are unique to Vascepa and extend beyond triglyceride reduction. If the FDA were to change this position, it could potentially have a negative impact on Amarin by making it easier for other products to achieve a cardiovascular risk reduction indication without the need in advance to conduct a long and expensive cardiovascular outcomes study.
Generic company competitors are seeking FDA approval of generic versions of Vascepa. We are now engaged in related patent litigation and could face other challenges to our exclusivity.
The FDCA, as amended by the Drug Price Competition and Patent Term Restoration Act of 1984, as amended, or the Hatch-Waxman Amendments, permits the FDA to approve ANDAs for generic versions of brand name drugs like Vascepa. We refer to the process of generic drug applications as the “ANDA process.” The ANDA process permits competitor companies to obtain marketing approval for a drug product with the same active ingredient, dosage form, strength, route of administration, and labeling as the approved brand name drug, but without having to conduct and submit clinical studies to establish the safety and efficacy of the proposed generic product. In place of such clinical studies, an ANDA applicant needs to submit data demonstrating that its product is bioequivalent to the brand name product, usually based on pharmacokinetic studies.
As an alternate path to FDA approval for modifications of products previously approved by the FDA, an applicant may submit a new drug application, or NDA, under Section 505(b)(2) of the FDCA (enacted as part of the Hatch-Waxman Amendments). This
34
statutory provision permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference from the owner of the data. The Hatch-Waxman Amendments permit the applicant to rely upon the FDA findings of safety and effectiveness of a drug that has obtained FDA approval based on preclinical or clinical studies conducted by others. In addition to relying on FDA prior findings of safety and effectiveness for a referenced drug product, the FDA may require companies to perform additional preclinical or clinical studies to support approval of the modification to the referenced product.
If an application for a generic version of a branded product or a Section 505(b)(2) application relies on a prior FDA finding of safety and effectiveness of a previously-approved product including an alternative strength thereof, the applicant is required to certify to the FDA concerning any patents listed for the referenced product in the FDA publication called “Approved Drug Products with Therapeutic Equivalence Evaluations,” otherwise known as the “Orange Book.” Specifically, the applicant must certify in the application that:
|
|
(I) |
there is no patent information listed for the reference drug; |
|
|
(II) |
the listed patent has expired for the reference drug; |
|
|
(III) |
the listed patent for the reference drug has not expired, but will expire on a particular date and approval is sought after patent expiration; or |
|
|
(IV) |
the listed patent for the reference drug is invalid, unenforceable, or will not be infringed by the manufacture, use or sale of the product for which the ANDA or 505(b)(2) NDA is submitted. |
The Hatch-Waxman Amendments require an applicant for a drug product that relies, in whole or in part, on the FDA’s prior approval of Vascepa, to notify us of its application, a “paragraph IV” notice, if the applicant is seeking to market its product prior to the expiration of the patents that both claim Vascepa and are listed in the Orange Book. A bona fide paragraph IV notice may not be given under the Hatch-Waxman Amendments until after the generic company receives from the FDA an acknowledgement letter stating that its ANDA is sufficiently complete to permit a substantive review.
The paragraph IV notice is required to contain a detailed factual and legal statement explaining the basis for the applicant’s opinion that the proposed product does not infringe our patents, that the relevant patents are invalid, or both. After receipt of a valid notice, the branded product manufacturer has the option of bringing a patent infringement suit in federal district court against any generic company seeking approval for its product within 45 days from the date of receipt of each notice. If such a suit is commenced within this 45-day period, the Hatch-Waxman Amendments provide for a 30-month stay on FDA’s ability to give final approval to the proposed generic product, which period begins on the date the paragraph IV notice is received. Generally, during a period of time in which generic applications may be submitted for a branded product based on a product’s regulatory exclusivity status, if no patents are listed in the Orange Book before the date on which a complete ANDA application for a product (excluding an amendment or supplement to the application) is submitted, an ANDA application could be approved by FDA without regard to a stay. For products entitled to five-year exclusivity status, the Hatch-Waxman Amendments provide that an ANDA application may be submitted after four years following FDA approval of the branded product if it contains a certification of patent invalidity or non-infringement to a patent listed in the Orange Book. In such a case, the 30-month stay runs from the end of the five-year exclusivity period. Statutory stays may be shortened or lengthened if either party fails to cooperate in the litigation and it may be terminated if the court decides the case in less than 30 months. If the litigation is resolved in favor of the ANDA applicant before the expiration of the 30-month period, the stay will be immediately lifted and the FDA’s review of the application may be completed. Such litigation is often time-consuming and costly, and may result in generic competition if such patents are not upheld or if the generic competitor is found not to infringe such patents.
In the first half of 2014, we received six paragraph IV notices notifying us of accepted ANDAs to the Vascepa 1-gram dose strength under the Hatch-Waxman Amendments. These ANDAs were submitted and accepted by FDA under the regulatory scheme adopted under the Hatch-Waxman Amendments based on the FDA’s determination that we were entitled to three, and not five-year exclusivity. As a result, from the first half of 2014 until June 2015, we were engaged in costly litigation with the ANDA applicants to protect our patent rights.
Based on the May 28, 2015, District of Columbia court order granting our motion for summary judgment in the new chemical entity, or NCE, litigation, on June 26, 2015, the parties to the related Vascepa patent litigation that followed acceptance by FDA of ANDAs to Vascepa based on a three-year regulatory exclusivity determination, agreed to a full stay of proceeding in that patent litigation.
Following the May 28, 2015 District of Columbia court order setting aside FDA’s denial of NCE exclusivity for Vascepa, FDA notified the ANDA filers that FDA had changed the status of their ANDAs to submitted, but no longer accepted, and notified ANDA filers that FDA had ceased review of the pending ANDAs. In rescinding acceptance of the ANDAs, the statutory basis for the patent litigation (accepted ANDAs) no longer existed. Thus, in July 2015, we moved to dismiss the pending patent infringement lawsuits against each of the Vascepa ANDA applicants in the U.S. District Court for the District of New Jersey.
35
On January 22, 2016, the U.S. District Court for the District of New Jersey granted our motion to dismiss all patent infringement litigation related to the 2014 acceptance by the FDA of ANDAs to Vascepa. An appeal of the court’s dismissal was filed by one ANDA filer and, after FDA’s May 2016 grant of Vascepa NCE exclusivity, that appeal was withdrawn by the ANDA filer. This dismissal and terminated appeal ended this patent litigation related to Vascepa.
On May 31, 2016, in a reversal that FDA and we view as consistent with the court’s May 28, 2015 summary judgment motion, FDA determined that Vascepa is eligible for five-year, NCE marketing exclusivity. This determination provides Vascepa with the benefits of NCE exclusivity afforded by statute. NCE exclusivity for Vascepa ran from its date of FDA approval on July 26, 2012 and extended until July 26, 2017. We believe the statutory NCE-related 30-month stay triggered by the 1-gram dose patent litigation following generic application submissions permitted on July 26, 2016 is scheduled to continue until January 26, 2020, seven-and-a-half years from FDA approval, unless such patent litigation is resolved against us sooner.
In September and October 2016, we received paragraph IV certification notices from four companies contending to varying degrees that certain of our patents are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of the 1-gram dose strength of Vascepa as described in those companies’ ANDAs. These certifications were expected given the eligibility for submission of ANDAs under the NCE regulatory structure, after the expiration of four years from the July 2012 approval of Vascepa.
We filed patent infringement lawsuits against three of these four ANDA applicants. In October 2016, Amarin filed a lawsuit against Roxane Laboratories, Inc. and related parties, collectively, Roxane, in the U.S. District Court for the District of Nevada. The case against Roxane is captioned Amarin Pharma, Inc. et al. v. Roxane Laboratories, Inc. et al., Civ. A. No. 2:16-cv-02525 (D. Nev.). According to a stipulation filed with the Nevada court, in December 2016, Roxane transferred its ANDA to West-Ward Pharmaceuticals International Limited, which then designated West-Ward Pharmaceuticals Corp. (or together with West-Ward Pharmaceuticals International Limited, West-Ward) as its agent for FDA communications. In view of the ANDA transfer, in February 2017, West-Ward replaced Roxane and related parties as Defendants in the above-referenced case. The case against West-Ward is now captioned Amarin Pharma, Inc. et al. v. West-Ward Pharmaceuticals Corp. et al., Civ. A. No. 2:16-cv-02525 (D. Nev.). In November 2016, Amarin filed a lawsuit against Dr. Reddy’s Laboratories, Inc. and Dr. Reddy’s Laboratories, Ltd., collectively, DRL, in the U.S. District Court for the District of Nevada. The case against DRL is captioned Amarin Pharma, Inc. et al. v. Dr. Reddy’s Laboratories, Inc. et al., Civ. A. No. 2:16-cv-02562 (D. Nev.). In November 2016, Amarin filed a lawsuit against Teva Pharmaceuticals USA, Inc. and Teva Pharmaceuticals Industries Limited, or collectively, Teva, in the U.S. District Court for the District of Nevada. The case against Teva was captioned Amarin Pharma, Inc. et al. v. Teva Pharmaceuticals USA, Inc. et al., Civ. A. No. 2:16-cv-02658. In all three lawsuits, we are seeking, among other remedies, an order enjoining each defendant from marketing generic versions of the 1-gram dose strength of Vascepa before the last to expire of the asserted patents in 2030. The three lawsuits have been consolidated for pretrial proceedings.
The fourth ANDA applicant referenced above is Apotex Inc., or Apotex, which sent us a paragraph IV certification notice in September 2016. The notice reflected that Apotex made a paragraph IV notice as to some, but not all, of the patents listed in the Orange Book for Vascepa. Because Apotex did not make a paragraph IV certification as to all listed patents, Apotex cannot market a generic version of Vascepa before the last to expire of the patents for which Apotex did not make a paragraph IV certification, which is in 2030. At a later date, Apotex may elect to amend its ANDA in order to make a paragraph IV certification as to additional listed patents. If and when Apotex does make such an amendment, it would be required to send Amarin an additional paragraph IV certification notice, and Amarin would then have the ability to file a lawsuit against Apotex pursuant to the Hatch-Waxman Amendments.
In October 2016, we introduced to the market a 0.5-gram dose strength of Vascepa. In August 2017, as anticipated, we received a paragraph IV certification notice from Teva contending that certain of our patents are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of the 0.5-gram dose strength of Vascepa, as described in the Teva ANDA. This Teva ANDA was filed as an amendment to the 1-gram Teva ANDA and is related to patents already at issue in the 1-gram Vascepa patent litigation. This certification followed the related listing in the Orange Book of patents associated with the 0.5-gram product in June 2017. This June 2017 listing was within the five-year, post NDA-approval period during which the Hatch-Waxman Amendments require a paragraph IV certification of patent invalidity or non-infringement under the Hatch-Waxman, five-year, NCE regulatory scheme. Accordingly, in October 2017, we filed a patent infringement lawsuit against Teva in the U.S. District Court for the District of Nevada. The case is captioned Amarin Pharma, Inc. et al. v. Teva Pharmaceuticals USA, Inc. et al., Civ. A. No. 2:17-cv-2641 (D. Nev.). In this lawsuit, we sought, among other remedies, an order enjoining Teva from marketing generic versions of the 0.5-gram dose strength of Vascepa before the last to expire of the asserted patents in 2030.
On May 24, 2018, we entered into a settlement agreement with Teva that resolves our ANDA patent litigation as it relates to Teva’s as amended ANDA for both the 1-gram and 0.5-gram dose strengths of Vascepa. As part of this settlement agreement, Teva may first begin selling its generic version of Vascepa in the United States on August 9, 2029, or earlier under certain customary circumstances, including commercial launch by another generic manufacturer under certain circumstances, in which event Teva would
36
pay us certain royalties on its generic Vascepa products. The ANDA patent litigation continues in the United States District Court for the District of Nevada with parties West-Ward and DRL.
In July 2018, as anticipated, we received a paragraph IV certification notice from DRL contending that certain of our patents are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of the 0.5-gram dose strength of Vascepa, as described in the DRL ANDA. This DRL ANDA was filed as an amendment to the 1-gram DRL ANDA and is related to patents already at issue in the 1-gram Vascepa patent litigation. This certification followed the related listing in the Orange Book of patents associated with the 0.5-gram product in June 2017. This June 2017 listing was within the five-year, post NDA-approval period during which the Hatch-Waxman Amendments require a paragraph IV certification of patent invalidity or non-infringement under the Hatch-Waxman, five-year, NCE regulatory scheme. Accordingly, in August 2018, we filed a patent infringement lawsuit against DRL in the U.S. District Court for the District of Nevada. The case is captioned Amarin Pharma, Inc. et al. v. Dr. Reddy’s Laboratories, Inc. et al., Civ. A. No. 2:18-cv-01596 (D. Nev.). In this lawsuit, we are seeking, among other remedies, an order enjoining DRL from marketing generic versions of the 0.5-gram dose strength of Vascepa before the last to expire of the asserted patents in 2030. In light of the overlap between the cases, DRL and Amarin have stipulated that the final judgment on the merits of the parties’ contentions in the consolidated 1-gram action shall also be binding in the 0.5-gram case.
We may also face challenges to the validity of our patents through a procedure known as inter partes review. Inter partes review is a trial proceeding conducted through the Patent Trial and Appeal Board, or PTAB, of the U.S. Patent and Trademark Office. Such a proceeding could be introduced against us within the statutory one-year window triggered by service of a complaint for infringement or at any time by an entity not served with a complaint. Such proceedings may review the patentability of one or more claims in a patent on specified substantive grounds such as allegations that a claim is obvious on the basis of certain prior art.
We intend to vigorously enforce our intellectual property rights relating to Vascepa, but we cannot predict the outcome of the pending lawsuits or any subsequently filed lawsuits or inter partes review.
If an ANDA filer meets the approval requirements for a generic version of Vascepa to the satisfaction of the FDA under its ANDA, FDA may grant tentative approval to the ANDA during a Hatch-Waxman 30-month stay period. A tentative approval is issued to an ANDA applicant when its application is approvable prior to the expiration of any exclusivities applicable to the branded, reference listed drug product. A tentative approval does not allow the applicant to market the generic drug product and postpones the final ANDA approval until any exclusivity protections, such as a 30-month stay, have expired. As a result of the statutory stays associated with the filing of these lawsuits under the Hatch-Waxman Amendments, we believe the FDA cannot grant final approval to West-Ward, DRL, or Teva’s respective ANDAs for the 1-gram strength of Vascepa before January 26, 2020, unless there is an earlier court decision holding that the subject patents are not infringed and/or are invalid.
If final approval is granted and an ANDA filer is able to supply the product in significant commercial quantities, the generic company could introduce a generic version of Vascepa. Any such introduction of a generic version of Vascepa would also be subject to current patent infringement claims including those being litigated in the above-detailed patent litigations, and any court order we may seek and be granted to prevent any such launch based on our patent claims prior to any adverse court judgment or PTAB finding against us.
Any generic market entry would limit our U.S. sales, which would have a significant adverse impact on our business and results of operations. In addition, even if a competitor’s effort to introduce a generic product is ultimately unsuccessful, the perception that such development is in progress and/or news related to such progress could materially affect the perceived value of our company and our stock price.
Vascepa’s five-year, NCE and related exclusivity benefits could be challenged by companies seeking to introduce generic versions of Vascepa.
The timelines and conditions under the ANDA process that permit the start of patent litigation and allow the FDA to approve generic versions of brand name drugs like Vascepa differ based on whether a drug receives three-year, or five-year, NCE marketing exclusivity. In May 2016, after significant litigation, FDA determined that Vascepa is eligible for NCE marketing exclusivity. Accordingly, we believe a related 30-month stay is currently in place with respect to our 1-gram dose strength of Vascepa that is scheduled to continue until January 26, 2020, seven-and-a-half years from FDA approval of Vascepa, unless related patent litigation is resolved against us sooner.
The FDA typically makes a determination on marketing exclusivity in connection with an NDA approval of a drug for a new indication. FDA marketing exclusivity is separate from, and in addition to, patent protection, trade secrets and manufacturing barriers to entry which could also help protect Vascepa against generic competition.
We applied to the FDA for five-year, NCE marketing exclusivity for Vascepa in connection with the NDA for our MARINE indication, which NDA was approved by the FDA on July 26, 2012. On February 21, 2014, in connection with the July 26, 2012
37
approval of the MARINE indication, the FDA denied a grant of five-year NCE marketing exclusivity to Vascepa and granted three-year marketing exclusivity. Under applicable regulations, such three-year exclusivity would have extended through July 25, 2015 and would have been supplemented by a 30-month stay triggered by patent litigation that would have extended into September 2016, unless such patent litigation was resolved against us sooner.
NCE marketing exclusivity precludes approval during the five-year exclusivity period of certain 505(b)(2) applications and ANDAs submitted by another company for another version of the drug. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. In such case, the pioneer drug company is afforded the benefit of a 30-month stay against the launch of such a competitive product that extends from the end of the five-year exclusivity period. A pioneer company could also be afforded extensions to the stay under applicable regulations, including a six-month pediatric exclusivity extension or a judicial extension if applicable requirements are met. A drug sponsor could also gain a form of marketing exclusivity under the Hatch-Waxman Amendments if such company can, under certain circumstances, complete a human clinical trial process and obtain regulatory approval of its product.
In contrast, a three-year period of exclusivity under the Hatch-Waxman Amendments is generally granted for a drug product that contains an active moiety that has been previously approved, such as when the application contains reports of new clinical investigations (other than bioavailability studies) conducted by the sponsor that were essential to approval of the application. Accordingly, we expect to receive three-year exclusivity in connection with any future regulatory approvals of Vascepa, such as an approval sought based on positive REDUCE-IT outcomes study results. Such three-year exclusivity protection precludes the FDA from approving a marketing application for an ANDA, a product candidate that the FDA views as having the same conditions of approval as Vascepa (for example, the same indication and/or other conditions of use), or a 505(b)(2) NDA submitted to the FDA with Vascepa as the reference product, for a period of three years from the date of FDA approval. The FDA may accept and commence review of such applications during the three-year exclusivity period. Such three-year exclusivity grant does not prevent a company from challenging the validity of patents at any time, subject to any prior four-year period pending from a grant of five-year exclusivity. This three-year form of exclusivity may also not prevent the FDA from approving an NDA that relies only on its own data to support the change or innovation.
On February 27, 2014, we sued the FDA in the U.S. District Court for the District of Columbia to challenge the agency’s denial of five-year NCE exclusivity for Vascepa, based on our reading of the relevant statute, our view of FDA’s inconsistency with its past actions in this area and the retroactive effect of what we believe is a new policy at FDA as it relates to our situation. On May 28, 2015, the court granted our motion for summary judgment. The decision vacated the FDA’s denial of our claim for such exclusivity and remanded to the FDA for proceedings consistent with the decision. On July 22, 2015, Watson Laboratories Inc., the purported first Vascepa ANDA filer, sought to intervene and appeal the court’s decision. We and FDA opposed this intervention effort. The applicable courts denied Watson the relief sought and appeal periods have expired.
On May 31, 2016, in a reversal that FDA and we view as consistent with the court’s May 28, 2015 summary judgment motion, FDA determined that Vascepa is eligible for five-year, NCE marketing exclusivity. We believe this determination provides Vascepa with the benefits of NCE exclusivity afforded by statute. NCE exclusivity for Vascepa ran from its date of FDA approval on July 26, 2012 and extended until July 26, 2017. We believe the statutory NCE-related 30-month stay triggered by the 1-gram dose patent litigation following generic application submissions permitted on July 26, 2016 is scheduled to continue until January 26, 2020, seven-and-a-half years from FDA approval, unless such patent litigation is resolved against us sooner.
It is possible that FDA’s NCE determination and related 30-month stay could be challenged by interested parties. If challenged, we plan to vigorously defend exclusivity for Vascepa. Any such challenge could have a negative impact on our company and create uncertainty around the continued benefits associated with exclusivity that we believe are applicable to us under the Hatch-Waxman Amendments.
Regulatory exclusivity is in addition to exclusivity afforded by issued patents related to Vascepa.
Vascepa is a prescription-only omega-3 fatty acid product. Omega-3 fatty acids are also marketed by other companies as non-prescription dietary supplements. As a result, Vascepa is subject to non-prescription competition and consumer substitution.
Our only product, Vascepa, is a prescription-only form of EPA, an omega-3 fatty acid in ethyl ester form. Mixtures of omega-3 fatty acids in triglyceride form are naturally occurring substances contained in various foods, including fatty fish. Omega-3 fatty acids are marketed by others in a number of chemical forms as non-prescription dietary supplements. We cannot be sure physicians will view the pharmaceutical grade purity and proven efficacy and safety of Vascepa as having a superior therapeutic profile to unproven and loosely regulated omega-3 fatty acid dietary supplements. In addition, the FDA has not yet enforced to the full extent of its regulatory authority what we view as illegal claims made by certain omega-3 fatty acid product manufacturers to the extent we believe appropriate under applicable law and regulations, for example, claims that certain of such chemically altered products are dietary supplements and that certain of such products reduce triglyceride levels or could reduce cardiovascular risk.
38
Also, for more than a decade now, subject to certain limitations, the FDA has expressly permitted dietary supplement manufacturers that sell supplements containing the omega-3 fatty acids EPA and/or DHA to make the following qualified health claim directly to consumers: Supportive but not conclusive research shows that consumption of EPA and DHA omega-3 fatty acids may reduce the risk of coronary heart disease. As a result of our First Amendment litigation and settlement, we may now make this claim to healthcare professionals subject to certain qualifications.
These factors enable dietary supplements to compete with Vascepa to a certain degree. Although we have taken steps to address these competitive issues, and plan to continue to do so vigorously, we may not be successful in such efforts. For example, on August 30, 2017, Amarin Pharma, Inc. and Amarin Pharmaceuticals Ireland Limited, each wholly-owned subsidiaries of Amarin Corporation plc, filed a lawsuit with the United States International Trade Commission, or the ITC, against manufacturers, importers, and distributors of products containing synthetically produced omega-3 products in ethyl ester or re-esterified triglyceride form that contain more EPA than DHA or any other single component for use in or as dietary supplements. The lawsuit sought an investigation by the ITC regarding potentially unfair methods of competition and unfair acts involving the importation and sale of articles in the United States that injure or threaten injury to a domestic industry. On October 27, 2017, the ITC determined to not institute our requested investigation. We are currently appealing this determination in federal court and plan to pursue it vigorously. We have also recently sued several omega-3 dietary supplement manufacturers for making claims that we believe make them unfairly competitive to Vascepa.
In addition, to the extent the net price of Vascepa after insurance and offered discounts is significantly higher than the prices of commercially available omega-3 fatty acids marketed by other companies as dietary supplements (through that lack of coverage by insurers or otherwise), physicians and pharmacists may recommend these commercial alternatives instead of writing or filling prescriptions for Vascepa or patients may elect on their own to take commercially available omega-3 fatty acids. Also, insurance plans may increasingly impose policies that favor supplement use over Vascepa. While Vascepa is highly price-competitive for patients generally, and in particular when covered by insurance—cheaper in many cases—any of these outcomes may adversely impact our results of operations by limiting how we price our product and limiting the revenue we receive from the sale of Vascepa due to reduced market acceptance.
We may not be successful in replacing our Vascepa co-promotion effort with Kowa Pharmaceuticals America, Inc. after it expired at the end of 2018.
In March 2014, we entered into a co-promotion agreement with Kowa Pharmaceuticals America, Inc. to co-promote Vascepa in the United States under which Kowa Pharmaceuticals America, Inc. co-promoted Vascepa in conjunction with its promotion of its primary product, a branded statin for patients with high cholesterol. Co-promotion under the agreement commenced in May 2014 and ceased at the end of 2018. We may seek to search for another commercialization partner, though there is no guarantee we would be successful in doing so. If we do not enter into a co-promotion agreement with an equally capable company or if our newly hired sales representatives are not effective as planned, our sales may be negatively impacted. If we elect to increase our expenditures to fund development or commercialization activities on our own, depending on Vascepa’s revenues, we may need to obtain additional capital, which may not be available to us on acceptable terms, or at all, or which may not be possible due to our other financing arrangements. If we do not generate sufficient funds from the sale of Vascepa or, to the extent needed to supplement funds generated from product revenue, cannot raise sufficient funds, we may not be able to devote resources sufficient to market and sell Vascepa on our own in a manner required to realize the full market potential of Vascepa.
The commercial value to us of current and sought marketing rights may be smaller than we anticipate.
There can be no assurance as to the adequacy for commercial success of the scope and breadth of the marketing rights we currently have or, if approved, an indication based on a successful outcome of the REDUCE-IT study. Even if we obtain marketing approval for additional indications, the FDA may impose restrictions on the product’s conditions for use, distribution or marketing and in some cases may impose ongoing requirements for post-market surveillance, post-approval studies or clinical trials. Also, the number of actual patients with conditions within the scope of our marketing efforts may be smaller than we anticipate. If any such marketing right or approved indication is narrower than we anticipate, the market potential for our product would suffer.
Our special protocol assessment, or SPA, agreement for ANCHOR was rescinded and our SPA agreement for REDUCE-IT is not a guarantee of FDA approval of Vascepa for proposed REDUCE-IT indications.
The REDUCE-IT trial was conducted pursuant to a SPA agreement, with the FDA, which means that the FDA agreed, based on the information we submitted to the agency, that the design and planned analysis of the trial was adequate to support use of the conducted study as the primary basis for approval with respect to effectiveness. A SPA agreement does not cover every aspect of clinical trial conduct and assessment. For example, secondary and/or tertiary endpoints, their ordering in the statistical hierarchy, their clinical significance, or whether any would yield results appropriate for labeling are considered review issues and are not intended to be a binding component of the REDUCE-IT SPA agreement. Further, matters such as endpoint adjudication procedures (including potential endpoint ascertainment, adjudication process and detailed definitions) were specified by FDA as issues to be reviewed by the
39
agency as part of a drug approval application. Consistent with the May 2016 FDA SPA draft guidance, FDA stated that the SPA agreement does not necessarily indicate the agency’s agreement with every detail of a protocol; instead, such an agreement indicates FDA’s concurrence with the elements critical to ensuring that the trial conducted under the protocol would have the potential to form the primary basis of an efficacy claim in a marketing application.
A SPA agreement is not a guarantee of approval. A SPA agreement is generally binding upon the FDA except in limited circumstances, such as if the FDA identifies a substantial scientific issue essential to determining safety or efficacy after the study begins, or if the study sponsor fails to follow the protocol that was agreed upon with the FDA. The FDA reserves the right of final determinations for approval based on its review of the entire data presented in a marketing application. The FDA previously rescinded our SPA agreement with the FDA for our ANCHOR trial because the FDA determined that a substantial scientific issue essential to determining the effectiveness of Vascepa in the studied population was identified after testing began. There can be no assurance that the FDA or the applicable regulatory authorities in other jurisdictions will not reach a similar conclusion with respect to the results of the REDUCE-IT trial or will not require additional studies by of Vascepa in additional patient populations.
The commercial value to us of sales of Vascepa outside the United States may be smaller than we anticipate.
There can be no assurance as to the adequacy for commercial success of Vascepa outside the United States. For example, even if we and Eddingpharm (Asia) Macao Commercial Offshore Limited, or Eddingpharm, obtain marketing approval in Mainland China, Hong Kong, Macau and Taiwan, or the China Territory, applicable regulatory agencies may impose restrictions on the product’s conditions for use, distribution or marketing and in some cases may impose ongoing requirements for post-market surveillance, post-approval studies or clinical trials. Also, there is a degree of unpredictability with regard to the eventual pricing and reimbursement levels of medications in markets outside the United States. If the pricing and reimbursement levels of Vascepa are lower than we anticipate, then affordability of, and market access to, Vascepa may be adversely affected and thus market potential in these territories would suffer. Furthermore, with regard to any indications for which we may gain approval in territories outside the United States, the number of actual patients with the condition included in such approved indication may be smaller than we anticipate. If any such approved indication is narrower than we anticipate, the market potential in these countries for our product would suffer.
Our products and marketing efforts are subject to extensive post-approval government regulation.
Once a product candidate receives FDA marketing approval, numerous post-approval requirements apply. Among other things, the holder of an approved NDA is subject to periodic and other monitoring and reporting obligations enforced by the FDA and other regulatory bodies, including obligations to monitor and report adverse events and instances of the failure of a product to meet the specifications in the approved application. Application holders must also submit advertising and other promotional material to regulatory authorities and report on ongoing clinical trials.
With respect to sales and marketing activities including direct-to-healthcare provider and direct-to-consumer advertising and promotional activities involving the internet, advertising and promotional materials must comply with FDA rules in addition to other applicable federal and local laws in the United States and in other countries. The result of our First Amendment litigation and settlement may cause the government to scrutinize our promotional efforts or otherwise monitor our business more closely. Industry-sponsored scientific and educational activities also must comply with FDA and other requirements. In the United States, the distribution of product samples to physicians must comply with the requirements of the U.S. Prescription Drug Marketing Act. Manufacturing facilities remain subject to FDA inspection and must continue to adhere to the FDA’s pharmaceutical current good manufacturing practice requirements, or cGMPs. Application holders must obtain FDA approval for product and manufacturing changes, depending on the nature of the change.
We also are subject to the federal transparency requirements under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively the ACA, enacted in March 2010, which require manufacturers of certain drugs, devices, biologics, and medical supplies to report to the Centers for Medicare & Medicaid Services, or CMS, information related to payments and other transfers of value to physicians and teaching hospitals and physician ownership and investment interests. We may also be subject, directly or indirectly through our customers and partners, to various fraud and abuse laws, including, without limitation, the U.S. Anti-Kickback Statute, U.S. False Claims Act, and similar state laws, which impact, among other things, our proposed sales, marketing, and scientific/educational grant programs. If we participate in the U.S. Medicaid Drug Rebate Program, the Federal Supply Schedule of the U.S. Department of Veterans Affairs, or other government drug programs, we will be subject to complex laws and regulations regarding reporting and payment obligations. We must also comply with requirements to collect and report adverse events and product complaints associated with our products. For example, in September 2014, we participated in a routine inspection from the FDA in which the FDA made observations on perceived deficiencies related to our processes for collection and processing of adverse events. We have responded to FDA with respect to these observations and continue to work with FDA to show that we have improved related systems and, given we received communication from the FDA that it considers this matter to be closed, we believe that we have demonstrated to FDA that we have adequately responded to these observations. Our activities are also subject to U.S. federal and state consumer protection and unfair competition laws, non-compliance with which could subject us to significant liability. Similar requirements exist in many of these areas in other countries.
40
Depending on the circumstances, failure to meet these post-approval requirements can result in criminal prosecution, fines or other penalties, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, or refusal to allow us to enter into supply contracts, including government contracts. We may also be held responsible for the non-compliance of our partners, such as Kowa Pharmaceuticals America, Inc. In addition, even if we comply with FDA and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA to modify or withdraw a product approval. Newly discovered or developed safety or effectiveness data may require changes to a drug’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Adverse regulatory action, whether pre- or post-approval, can potentially lead to product liability claims and increase our product liability exposure. We must also compete against other products in qualifying for coverage and reimbursement under applicable third-party payment and insurance programs. In addition, all of the above factors may also apply to any regulatory approval for Vascepa obtained in territories outside the United States. Given our inexperience with marketing and commercializing products outside the United States, we will need to rely on third parties, such as Eddingpharm in China, to assist us in dealing with any such issues.
Legislative or regulatory reform of the healthcare system in the United States and foreign jurisdictions may affect our ability to profitably sell Vascepa.
Our ability to commercialize our future products successfully, alone or with collaborators, will depend in part on the extent to which coverage and reimbursement for the products will be available from government and health administration authorities, private health insurers and other third-party payors. The continuing efforts of the U.S. and foreign governments, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce healthcare costs may adversely affect our ability to set prices for our products which we believe are fair, and our ability to generate revenues and achieve and maintain profitability.
Specifically, in both the United States and some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. For example, beginning April 1, 2013, Medicare payments for all items and services, including drugs and biologics, were reduced by 2% under the sequestration (i.e., automatic spending reductions) required by the Budget Control Act of 2011, as amended by the American Taxpayer Relief Act of 2012. Subsequent legislation extended the 2% reduction, on average, to 2027. These cuts reduce reimbursement payments related to our products, which could potentially negatively impact our revenue. Also for example, the ACA substantially changed the way healthcare is financed by both governmental and private insurers. Among other cost-containment measures, ACA establishes:
|
|
• |
an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents; |
|
|
• |
a new Medicare Part D coverage gap discount program, in which pharmaceutical manufacturers who wish to have their drugs covered under Part D must offer discounts to eligible beneficiaries during their coverage gap period; |
|
|
• |
a new formula that increases the rebates a manufacturer must pay under the Medicaid Drug Rebate Program; and |
|
|
• |
new policies or laws affecting Vascepa sales, such as state and federal efforts to affect drug pricing and provide healthcare coverage that includes reimbursement for prescription drugs. |
There has been increasing legislative and enforcement interest in the United States with respect to specialty drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. At the federal level, the Trump administration’s budget proposal for fiscal year 2019 contains further drug price control measures that could be enacted during the 2019 budget process or in other future legislation, including, for example, measures to permit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid, and to eliminate cost sharing for generic drugs for low-income patients. Additionally, the Trump administration released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. The U.S. Department of Health and Human Services, or HHS, has already started the process of soliciting feedback on some of these measures and, at the same time, is immediately implementing others under its existing authority. For example, in September 2018, CMS announced that it will allow Medicare Advantage Plans the option to use step therapy for Part B drugs beginning January 1, 2019, and in October 2018, CMS proposed a new rule that would require direct-to-consumer television advertisements of prescription drugs and biological products, for which payment is available through or under Medicare or Medicaid, to include in the advertisement the Wholesale Acquisition Cost, or list price, of that drug or biological product. Although a number of these, and other proposed measures will require authorization through additional legislation to become effective, Congress and the Trump administration have each indicated
41
that it will continue to seek new legislative and/or administrative measures to control drug costs. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. For example, the State of California enacted legislation that requires notice for exceeding specified limits on annual drug price increases and other legislation that seeks to limit the use of co-pay cards in certain situations.
In addition, it is time-consuming and expensive for us to go through the process of seeking coverage and reimbursement from Medicare and private payors. Our products may not be considered cost effective, and government and third-party private health insurance coverage and reimbursement may not be available to patients for any of our future products or sufficient to allow us to sell our products on a competitive and profitable basis. Our results of operations could be adversely affected by ACA and by other healthcare reforms that may be enacted or adopted in the future. In addition, increasing emphasis on managed care in the United States will continue to put pressure on the pricing of pharmaceutical products. For example, proposals are being considered to expand the use of dietary supplements in addition to or in place of drugs in government and private payor plans. In addition, cost control initiatives could decrease the price that we or any potential collaborators could receive for any of our future products and could adversely affect our profitability.
In some foreign countries, including major markets in the European Union and Japan, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take 6 to 12 months or longer after the receipt of regulatory marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a pharmacoeconomic study that compares the cost-effectiveness of Vascepa to other available therapies. Such pharmacoeconomic studies can be costly and the results uncertain. Our business could be harmed if reimbursement of our products is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels.
If we fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate program or other governmental pricing programs, we could be subject to additional reimbursement requirements, penalties, sanctions and fines, which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We participate in the Medicaid Drug Rebate program, the 340B drug pricing program, and the U.S. Department of Veterans Affairs, or VA, Federal Supply Schedule, or FSS, pricing program. Under the Medicaid Drug Rebate program, we are required to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under Medicaid and Medicare Part B. Those rebates are based on pricing data reported by us on a monthly and quarterly basis to CMS, the federal agency that administers the Medicaid Drug Rebate program. These data include the average manufacturer price and, in the case of innovator products, the best price for each drug which, in general, represents the lowest price available from the manufacturer to any entity in the U.S. in any pricing structure, calculated to include all sales and associated rebates, discounts and other price concessions. Our failure to comply with these price reporting and rebate payment obligations could negatively impact our financial results.
The ACA made significant changes to the Medicaid Drug Rebate program. CMS issued a final regulation, which became effective on April 1, 2016, to implement the changes to the Medicaid Drug Rebate program under the ACA. The issuance of the final regulation has increased and will continue to increase our costs and the complexity of compliance, has been and will continue to be time-consuming to implement, and could have a material adverse effect on our results of operations, particularly if CMS challenges the approach we take in our implementation of the final regulation.
Federal law requires that any company that participates in the Medicaid Drug Rebate program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B program requires participating manufacturers to agree to charge statutorily defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients. The 340B ceiling price is calculated using a statutory formula based on the average manufacturer price and Medicaid rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate program, and in general, products subject to Medicaid price reporting and rebate liability are also subject to the 340B ceiling price calculation and discount requirement. Any additional future changes to the definition of average manufacturer price and the Medicaid rebate amount under the ACA, other legislation, or in regulation could affect our 340B ceiling price calculations and negatively impact our results of operations.
The Health Resources and Services Administration, or HRSA, which administers the 340B program, issued a final regulation regarding the calculation of the 340B ceiling price and the imposition of civil monetary penalties on manufacturers that knowingly and intentionally overcharge covered entities, which became effective on January 1, 2019. It is currently unclear how HRSA will apply its enforcement authority under the new regulation. HRSA also is implementing a ceiling price reporting requirement related to the 340B
42
program during the first quarter of 2019, pursuant to which we are required to report our 340B ceiling prices to HRSA on a quarterly basis. Implementation of the civil monetary penalties regulation and the issuance of any other final regulations and guidance could affect our obligations under the 340B program in ways we cannot anticipate. In addition, legislation may be introduced that, if passed, would further expand the 340B program to additional covered entities or would require participating manufacturers to agree to provide 340B discounted pricing on drugs used in the inpatient setting.
Pricing and rebate calculations vary across products and programs, are complex, and are often subject to interpretation by us, governmental or regulatory agencies and the courts. In the case of our Medicaid pricing data, if we become aware that our reporting for a prior quarter was incorrect, or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected data for up to three years after those data originally were due. Such restatements and recalculations increase our costs for complying with the laws and regulations governing the Medicaid Drug Rebate program and could result in an overage or underage in our rebate liability for past quarters. Price recalculations also may affect the ceiling price at which we are required to offer our products under the 340B program or could require us to issue refunds to 340B covered entities.
Significant civil monetary penalties can be applied if we are found to have knowingly submitted any false pricing information to CMS, or if we fail to submit the required price data on a timely basis. Such conduct also could be grounds for CMS to terminate our Medicaid drug rebate agreement, in which case federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs. Significant civil monetary penalties also can be applied if we are found to have knowingly and intentionally charged 340B covered entities more than the statutorily mandated ceiling price. We cannot assure you that our submissions will not be found by CMS or HRSA to be incomplete or incorrect.
In order to be eligible to have our products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, we participate in the U.S. Department of Veterans Affairs, or VA, Federal Supply Schedule, or FSS, pricing program. As part of this program, we are obligated to make our products available for procurement on an FSS contract under which we must comply with standard government terms and conditions and charge a price that is no higher than the statutory Federal Ceiling Price, or FCP, to four federal agencies (VA, U.S. Department of Defense, or DOD, Public Health Service, and U.S. Coast Guard). The FCP is based on the Non-Federal Average Manufacturer Price, or Non-FAMP, which we calculate and report to the VA on a quarterly and annual basis. Pursuant to applicable law, knowing provision of false information in connection with a Non-FAMP filing can subject a manufacturer to significant penalties for each item of false information. These obligations also contain extensive disclosure and certification requirements.
We also participate in the Tricare Retail Pharmacy program, under which we pay quarterly rebates on utilization of innovator products that are dispensed through the Tricare Retail Pharmacy network to Tricare beneficiaries. The rebates are calculated as the difference between the annual Non-FAMP and FCP. We are required to list our covered products on a Tricare Agreement in order for these products to be eligible for DOD formulary inclusion. If we overcharge the government in connection with our FSS contract or Tricare Agreement, whether due to a misstated FCP or otherwise, we are required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the False Claims Act and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Changes in reimbursement procedures by government and other third-party payors may limit our ability to market and sell our approved drugs. These changes could have a material adverse effect on our business and financial condition.
In the U.S. and abroad, sales of pharmaceutical drugs are dependent, in part, on the availability of reimbursement to the consumer from third-party payors, such as government and private insurance plans. Third-party payors are increasingly challenging the prices charged for medical products and services. Some third-party payor benefit packages restrict reimbursement, charge copayments to patients, or do not provide coverage for specific drugs or drug classes.
In addition, certain healthcare providers are moving toward a managed care system in which such providers contract to provide comprehensive healthcare services, including prescription drugs, for a fixed cost per person. We are unable to predict the reimbursement policies employed by third-party healthcare payors.
43
We expect to experience pricing and reimbursement pressures in connection with the sale of our products due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative proposals. In addition, we may confront limitations in insurance coverage for our products. If we fail to successfully secure and maintain reimbursement coverage for our approved drugs or are significantly delayed in doing so, we may have difficulty achieving market acceptance of our approved drugs and investigational drug candidates for which we obtain approval, and our business may be harmed. Congress has enacted healthcare reform and may enact further reform, which could adversely affect the pharmaceutical industry as a whole, and therefore could have a material adverse effect on our business.
Ongoing healthcare legislative and regulatory reform measures may have a material adverse effect on our business and results of operations.
Some of the provisions of the ACA have yet to be fully implemented, and certain provisions have been subject to judicial and Congressional challenges. In addition, there have been efforts by the Trump administration to repeal or replace certain aspects of the ACA and to alter the implementation of the ACA and related laws. Since January 2017, the Trump administration has signed two Executive Orders designed to delay the implementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by the ACA. One Executive Order directs federal agencies with authorities and responsibilities under the ACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the ACA that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. The second Executive Order terminates the cost-sharing subsidies that reimburse insurers under the ACA. Nineteen state Attorneys General filed suit to stop the administration from terminating the subsidies, but on July 18, 2018, the U.S. District Court for the Northern District of California dismissed the case without prejudice. Further, on June 14, 2018, U.S. Court of Appeals for the Federal Circuit ruled that, due to Congressional appropriations riders that prohibited the Department of Health and Human Services (HHS) from paying out more in risk corridor payments than it collected, HHS was not required to pay more than $12 billion in ACA risk corridor payments owed to insurers under the risk corridor formula. On November 6, 2018, the Federal Circuit declined to rehear the case en banc. Insurers have appealed this ruling to the Supreme Court.
Moreover, the Tax Cuts and Jobs Act enacted on December 22, 2017, eliminated the tax-based shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code of 1986, commonly referred to as the “individual mandate,” effective January 1, 2019. On January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain ACA-mandated fees, including the so-called “Cadillac” tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices. The Bipartisan Budget Act of 2018, or the BBA, among other things, amends the ACA, effective January 1, 2019, to reduce the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole” by raising the manufacturer discount under the Medicare Part D coverage gap discount program to 70%.
In July 2018, the Centers for Medicare and Medicaid Services, or CMS, published a final rule permitting certain further collections and payments to and from certain ACA qualified health plans and health insurance issuers under the ACA risk adjustment program in response to the outcome of federal district court litigation regarding the method CMS uses to determine this risk adjustment. In addition, CMS has published a final rule that gives states greater flexibility, starting in 2020, in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing the essential health benefits required under the ACA for plans sold through such marketplaces. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, or the Texas District Court Judge, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the Tax Cuts and Jobs Act of 2017, the remaining provisions of the ACA are invalid as well. This ruling is under appeal and stayed pending appeal. While the Trump Administration and CMS have both stated that the ruling will have no effect while this appeal is pending, it is unclear how this decision, subsequent appeals, and other efforts to invalidate the ACA or portions thereof will impact the ACA, its implementation, and our business.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. In August 2011, President Obama signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction, which triggered the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of, on average, 2% per fiscal year through 2025 unless Congress takes additional action. These reductions were extended through 2027 under the BBA. In January 2013, the American Taxpayer Relief Act of 2012, among other things, further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
We expect that the healthcare reform measures that have been adopted and may be adopted in the future, may result in more rigorous coverage criteria, and new payment methodologies, and in additional downward pressure on coverage and payment and the
44
price that we receive for any approved product, and could seriously harm our future revenues. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private third-party payors.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our product. Such reforms could have an adverse effect on anticipated revenue from product candidates that we may successfully develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop product candidates.
Failure to comply with health and data protection laws and regulations could lead to government enforcement actions (which could include civil or criminal penalties), private litigation, and/or adverse publicity and could negatively affect our operating results and business.
We and any potential collaborators may be subject to federal, state, and foreign data protection laws and regulations (i.e., laws and regulations that address privacy and data security). In the United States, numerous federal and state laws and regulations, including federal health information privacy laws, state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), that govern the collection, use, disclosure and protection of health-related and other personal information could apply to our operations or the operations of our collaborators. In addition, we may obtain health information from third parties (including research institutions from which we obtain clinical trial data) that are subject to privacy and security requirements under HIPAA. Although we are not directly subject to HIPAA – other than with respect to providing certain employee benefits – we could potentially be subject to criminal penalties if we, our affiliates, or our agents knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. In addition, state laws govern the privacy and security of health information in specified circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
Compliance with U.S. and international data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Failure to comply with these laws and regulations could result in government enforcement actions (which could include civil, criminal and administrative penalties), private litigation, and/or adverse publicity and could negatively affect our operating results and business. Moreover, clinical trial subjects, employees and other individuals about whom we or our potential collaborators obtain personal information, as well as the providers who share this information with us, may limit our ability to collect, use and disclose the information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
European data collection is governed by restrictive regulations governing the use, processing and cross-border transfer of personal information.
In the event we decide to conduct clinical trials in the European Union, or EU, we may be subject to additional privacy restrictions. The collection and use of personal health data in the EU is governed by the provisions of the General Data Protection Regulation, or GDPR. This directive imposes several requirements relating to the consent of the individuals to whom the personal data relates, the information provided to the individuals, notification of data processing obligations to the competent national data protection authorities and the security and confidentiality of the personal data. The GDPR also imposes strict rules on the transfer of personal data out of the European Union to the United States. Failure to comply with the requirements of the Data Protection Directive, the GDPR, and the related national data protection laws of the European Union Member States may result in fines and other administrative penalties. The GDPR introduces new data protection requirements in the European Union and substantial fines for breaches of the data protection rules. The GDPR regulations may impose additional responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with these and/or new data protection rules. This may be onerous and adversely affect our business, financial condition, prospects and results of operations.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products. If we or our partners are found to have improperly promoted uses, efficacy or safety of Vascepa, we may become subject to significant fines and other liability. The government may seek to find means to prevent our promotion of truthful and non-misleading information beyond the current court ruling and litigation settlement.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products. In particular, in general, the U.S. government’s position has been that a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. Even though we received FDA marketing approval for Vascepa for the MARINE indication and we believe the First Amendment court ruling and litigation settlement affords us a degree of protection for
45
other promotional efforts, physicians may still prescribe Vascepa to their patients for use in the treatment of conditions that are not included as part of the indication statement in our FDA-approved Vascepa label or our settlement. If we are found to have promoted Vascepa outside the terms of the litigation settlement or in violation of what federal or state government may determine to be acceptable, we may become subject to significant government fines and other related liability, such as under the FDCA, the False Claims Act, or other theories of liability. Government may also seek to hold us responsible for the non-compliance of our former co-promotion partner, Kowa Pharmaceuticals America, Inc., or our commercialization partners outside the United States. For example, the Federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed.
In addition, incentives exist under applicable laws that encourage competitors, employees and physicians to report violations of rules governing promotional activities for pharmaceutical products. These incentives could lead to so-called “whistleblower lawsuits” as part of which such persons seek to collect a portion of moneys allegedly overbilled to government agencies due to, for example, promotion of pharmaceutical products beyond labeled claims. These incentives could also lead to suits that we have mischaracterized a competitor’s product in the marketplace and we may, as a result, be sued for alleged damages to our competitors. Such lawsuits, whether with or without merit, are typically time-consuming and costly to defend. Such suits may also result in related shareholder lawsuits, which are also costly to defend.
Even though we have a final settlement in our litigation related to promotion beyond FDA-approved labeling, our promotion would still be subject to a high, perhaps abnormally high, degree of scrutiny to ensure that our promotion remains within the permitted scope. Likewise, federal or state government may seek to find other means to prevent our promotion of truthful and non-misleading information.
We may not be successful in developing or marketing future products if we cannot meet the extensive regulatory requirements of the FDA and other regulatory agencies for quality, safety and efficacy.
The success of our research and development efforts is dependent in part upon our ability, and the ability of our partners or potential partners, to meet regulatory requirements in the jurisdictions where we or our partners or potential partners ultimately intend to sell such products once approved. The development, manufacture and marketing of pharmaceutical products are subject to extensive regulation by governmental authorities in the United States and elsewhere. In the United States, the FDA generally requires preclinical testing and clinical trials of each drug to establish its safety and efficacy and extensive pharmaceutical development to ensure its quality before its introduction into the market. Regulatory authorities in other jurisdictions impose similar requirements. The process of obtaining regulatory approvals is lengthy and expensive and the issuance of such approvals is uncertain. The commencement and rate of completion of clinical trials and the timing of obtaining marketing approval from regulatory authorities may be delayed by many factors, including but not limited to:
|
|
• |
the lack of efficacy during clinical trials; |
|
|
• |
the inability to manufacture sufficient quantities of qualified materials under cGMPs for use in clinical trials; |
|
|
• |
slower than expected rates of patient recruitment; |
|
|
• |
the inability to observe patients adequately after treatment; |
|
|
• |
changes in regulatory requirements for clinical trials or preclinical studies; |
|
|
• |
the emergence of unforeseen safety issues in clinical trials or preclinical studies; |
|
|
• |
delay, suspension, or termination of a trial by the institutional review board responsible for overseeing the study at a particular study site; |
|
|
• |
unanticipated changes to the requirements imposed by regulatory authorities on the extent, nature or timing of studies to be conducted on quality, safety and efficacy; |
|
|
• |
government or regulatory delays or “clinical holds” requiring suspension or termination of a trial; and |
|
|
• |
political instability affecting our clinical trial sites. |
Even if we obtain positive results from early stage preclinical studies or clinical trials, we may not achieve the same success in future trials. Clinical trials that we or potential partners conduct may not provide sufficient safety and efficacy data to obtain the requisite regulatory approvals for product candidates. The failure of clinical trials to demonstrate safety and efficacy for our desired indications could harm the development of that product candidate as well as other product candidates, and our business and results of operations would suffer. For example, the efficacy results of our Vascepa Phase 3 clinical trials for the treatment of Huntington’s disease were negative. As a result, we stopped development of that product candidate, revised our clinical strategy and shifted our focus to develop Vascepa for use in the treatment of cardiovascular disease. Questions can also arise on the quality of study data or its reliability. For example, during the public advisory committee meeting held by FDA as part of its review of our ANCHOR sNDA, a
46
discussion regarding observed, nominally statistically significant changes from baseline in an adverse direction, while on background statin therapy, in certain lipid parameters, including triglycerides, in the placebo group, raised questions about the possibility that the mineral oil placebo used in the ANCHOR trial (and in the REDUCE-IT trial) might not be biologically inert and might be viewed as artificially exaggerating the clinical effect of Vascepa when measured against placebo in the ANCHOR trial. Ultimately, no strong evidence for biological activity of mineral oil was identified by the FDA before its approval of Vascepa after review of the MARINE and ANCHOR trials and consideration of other data regarding mineral oil. It was ultimately concluded that the between-group differences likely provided the most appropriate descriptions of the treatment effect of Vascepa and that whatever factor(s) led to the within-group changes over time in the placebo group were likely randomly distributed to all treatment groups. Thus, the FDA approved Vascepa for use in the MARINE indication in July 2012, FDA did not dispute the veracity of the ANCHOR trial data and, in connection with the March 2016 agreement we reached with the FDA allowing us to promote the results of the ANCHOR study, the FDA did not seek to require that we include any qualification related to this earlier question regarding the mineral oil placebo. The FDA, early on in the course of the REDUCE-IT trial, directed the DMC for REDUCE-IT to periodically review unblinded lipid data to monitor for signals that the placebo might not be inert. After each such quarterly unblinded safety analysis and review meeting to date, the DMC recommended to continue the REDUCE-IT study as planned. Each of these DMC recommendations has been shared with FDA. This matter illustrates that concerns such as this may arise in the future that could affect our product development, regulatory review or the public perception of our products and our future prospects, including REDUCE-IT results.
Any approvals that are obtained may be limited in scope, may require additional post-approval studies or may require the addition of labeling statements focusing on product safety that could affect the commercial potential for our product candidates. Any of these or similar circumstances could adversely affect our ability to gain approval for new indications and affect revenues from the sale of our products. Even in circumstances where products are approved by a regulatory body for sale, the regulatory or legal requirements may change over time, or new safety or efficacy information may be identified concerning a product, which may lead to the withdrawal of a product from the market or similar use restrictions. The discovery of previously unknown problems with a clinical trial or product, or in connection with the manufacturer of products, may result in regulatory issues that prevent proposed future approvals of a product and/or restrictions on that product or manufacturer, including withdrawal of an indication or the product from the market, which would have a negative impact on our potential revenue stream.
As we continue to evolve from a company primarily involved in research and development to a company also focused on establishing an infrastructure for commercializing Vascepa, we may encounter difficulties in managing our growth and expanding our operations successfully.
The process of establishing a commercial infrastructure is difficult, expensive and time-consuming. Prior to REDUCE-IT topline results announcement in September 2018, our sales team consisted of approximately 170 sales professionals, including sales representatives and their managers. We have recently increased the size of our sales force approximately 440 sales professionals, including approximately 400 sales representatives, in the United States and are expanding our promotion of Vascepa. This sales team promotes Vascepa to a limited group of physicians and other healthcare professionals in select geographies in the United States. Even after planned expansion, this sales team is not large enough to call upon all physicians.
In addition to sales force expansion in the United States, Amarin plans to work with its international partners to support regulatory efforts outside the United States based on REDUCE-IT results. As our operations expand with the anticipated growth of our product sales, we expect that we will need to manage additional relationships with various collaborative partners, suppliers and other third parties. Future growth will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain and integrate additional employees. Our future financial performance and our ability to commercialize Vascepa and to compete effectively will depend, in part, on our ability to manage our future growth effectively. To that end, we must be able to manage our development efforts effectively, and hire, train, integrate and retain additional management, administrative and sales and marketing personnel. We may not be able to accomplish these tasks, and our failure to accomplish any of them could prevent us from successfully growing our company.
Risks Related to Our Reliance on Third Parties
Our supply of product for the commercial market and clinical trials is dependent upon relationships with third-party manufacturers and suppliers.
We have no in-house manufacturing capacity and rely on contract manufacturers for our clinical and commercial product supply. We cannot ensure that we will successfully manufacture any product we may develop, either independently or under manufacturing arrangements, if any, with our third-party manufacturers. Moreover, if our manufacturers should cease doing business with us or experience delays, shortages of supply or excessive demands on their capacity, we may not be able to obtain adequate quantities of product in a timely manner, or at all.
Any manufacturing problem, natural disaster affecting manufacturing facilities, government action, or the loss of a contract manufacturer could be disruptive to our operations and result in lost sales. Any reliance on suppliers may involve several risks,
47
including a potential inability to obtain critical materials and reduced control over production costs, delivery schedules, reliability and quality. Any unanticipated disruption to future contract manufacture caused by problems at suppliers could delay shipment of products, increase our cost of goods sold and/or result in lost sales. If our suppliers were unable to supply us with adequate volumes of active pharmaceutical ingredient (drug substance) or encapsulated bulk product (drug product), it would have a material adverse effect on our ability to continue to commercialize Vascepa.
We have contractual freedom to source the API for Vascepa and to procure other services supporting our supply chain. We have entered into supply agreements with multiple suppliers who also rely on other third-party suppliers to manufacture the API and other elements necessary for the sale of Vascepa. Our strategy in sourcing API and other components in our supply chain from multiple suppliers has been to expand manufacturing capacity, maintain competitive advantages, and mitigate the risk of reliance on any single supplier.
Expanding manufacturing capacity and qualifying such capacity is complex and subject to numerous regulations and other operational challenges. The resources of our suppliers vary and are limited; costs associated with projected expansion and qualification can be significant. There can be no assurance that the expansion plans of any of our suppliers will be successful. Our aggregate capacity to produce API is dependent upon the continued qualification of our API suppliers and, depending on the ability of existing suppliers to meet our supply demands, potentially the qualifications of new suppliers. Each of our API suppliers has outlined plans for potential further capacity expansion. If no additional API supplier is approved by the FDA as part of an sNDA, our API supply will be limited to the API we purchase from previously approved suppliers. If our third-party manufacturing capacity is not expanded and/or compliant with applicable regulatory requirements, we may not be able to supply sufficient quantities of Vascepa to meet anticipated demand. We cannot guarantee that we can contract with any future manufacturer on acceptable terms or that any such alternative supplier will not require capital investment from us in order for them to meet our requirements. Alternatively, our purchase of supply may exceed actual demand for Vascepa.
There can be no guarantee that current suppliers and future suppliers with which we have contracted to encapsulate API will be continually qualified to manufacture the product to our specifications or that current and any future suppliers will have the manufacturing capacity to meet anticipated demand for Vascepa.
We may purchase too much or not enough supply to satisfy actual demand, which could have a material adverse effect on our financial results and financial condition.
Certain of our agreements with our suppliers include minimum purchase obligations and limited exclusivity provisions. These purchases are generally made on the basis of rolling twelve-month forecasts which in part are binding on us and the balance of which are subject to adjustment by us subject to certain limitations. Certain of our agreements also include contractual minimum purchase commitments regardless of the rolling twelve-month forecasts. We may not purchase sufficient quantities of Vascepa to meet actual demand or our purchase of supply may exceed actual demand. In either case, such event could have a material adverse effect on our financial results and financial condition.
The manufacture, packaging and distribution of pharmaceutical products such as Vascepa are subject to FDA regulations and those of similar foreign regulatory bodies. If we or our third-party manufacturers fail to satisfy these requirements, our product development and commercialization efforts may be materially harmed.
The manufacture, packaging and distribution of pharmaceutical products, such as Vascepa, are regulated by the FDA and similar foreign regulatory bodies and must be conducted in accordance with the FDA’s pharmaceutical current good manufacturing practices, or cGMPs, and comparable requirements of foreign regulatory bodies. There are a limited number of manufacturers that operate under these cGMPs and International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, or ICH, regulations and guidelines who are both capable of manufacturing Vascepa and willing to do so. Failure by us or our third-party manufacturers to comply with applicable regulations, requirements, or guidelines could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our products, delays, suspension or withdrawal of approvals, license revocation, seizures or voluntary recalls of product, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect our business. If we are not able to manufacture Vascepa to required specifications through our current and potential API suppliers, we may be delayed in successfully supplying the product to meet anticipated demand and our anticipated future revenues and financial results may be materially adversely affected.
Changes in the manufacturing process or procedure, including a change in the location where the product is manufactured or a change of a third-party manufacturer, may require prior FDA review and pre-approval of the manufacturing process and procedures in accordance with the FDA’s cGMPs. Any new facility may be subject to a pre-approval inspection by the FDA and would again require us to demonstrate product comparability to the FDA. There are comparable foreign requirements under ICH guidelines. This review may be costly and time consuming and could delay or prevent the launch of a product.
48
Furthermore, the FDA and foreign regulatory agencies require that we be able to consistently produce the API and the finished product in commercial quantities and of specified quality on a repeated basis, including demonstrated product stability, and document our ability to do so. This requirement is referred to as process validation. This includes stability testing, measurement of impurities and testing of other product specifications by validated test methods. If the FDA does not consider the result of the process validation or required testing to be satisfactory, the commercial supply of Vascepa may be delayed, or we may not be able to supply sufficient quantities of Vascepa to meet anticipated demand.
The FDA and similar foreign regulatory bodies may also implement new requirements, or change their interpretation and enforcement of existing requirements, for manufacture, packaging or testing of products at any time. If we or our approved suppliers are unable to comply, we may be subject to regulatory, civil actions or penalties which could significantly and adversely affect our business.
Our commercialization of Vascepa outside the United States is substantially dependent on third parties.
We have expanded our Vascepa commercialization activities outside of the United States through several contractual arrangements in territories including China, the Middle East, North Africa and Canada. We continue to assess other opportunities to develop Vascepa commercialization outside of the United States through similar arrangements.
In February 2015, we entered into a Development, Commercialization and Supply Agreement, or the DCS Agreement, with Eddingpharm related to the development and commercialization of Vascepa in the China Territory. Under the DCS Agreement, Eddingpharm is responsible for development and commercialization activities in the China Territory and associated expenses. Additionally, Eddingpharm is required to conduct clinical trials in the China Territory to secure regulatory approval in certain territories. For example, in December 2017, Eddingpharm commenced a pivotal clinical trial aimed to support the regulatory approval of the first indication of Vascepa in a patient population with severe hypertriglyceridemia in Mainland China. Additional clinical development efforts may be necessary in this market. Significant commercialization of Vascepa in the China Territory is several years away, if at all. If Eddingpharm is not able to effectively develop and commercialize Vascepa in the China Territory, we may not be able to generate revenue from the DCS Agreement resulting from the sale of Vascepa in the China Territory.
In March 2016, we entered into an agreement with Biologix FZCo, or Biologix, to register and commercialize Vascepa in several Middle Eastern and North African countries. Under the terms of the distribution agreement, we granted to Biologix a non-exclusive license to use our trademarks in connection with the importation, distribution, promotion, marketing and sale of Vascepa in the Middle East and North Africa territory. Commercialization across the Middle East and North Africa is several years away, if at all, in the most commercially significant territories and subject to similar risks as in the China Territory.
In September 2017, we entered into an agreement with HLS Therapeutics Inc., or HLS, to register, commercialize and distribute Vascepa in Canada. Under the agreement, HLS is responsible for regulatory and commercialization activities and associated costs. Amarin is responsible for providing assistance towards local filings, supplying finished product under negotiated supply terms, maintaining intellectual property, and continuing the development and funding of REDUCE-IT-related activities. Significant commercialization of Vascepa in Canada is several years away, if at all. If HLS Therapeutics is not able to effectively register and commercialize Vascepa in Canada, we may not be able to generate revenue from the agreement as a result of the sale of Vascepa in Canada.
We have limited experience working with partners outside the United States to develop and market our products in non-U.S. jurisdictions. In order for our partners to market and sell Vascepa in any country outside of the United States for any indication, it will be necessary to obtain regulatory approval from the appropriate regulatory authorities. The requirements and timing for regulatory approval, which may include conducting clinical trials, vary widely from country to country and may in some cases be different than or more rigorous than requirements in the United States. Any failure by us or our partners to obtain approval for Vascepa in non-U.S. jurisdictions in a timely manner may limit the commercial success of Vascepa and our ability to grow our revenues.
Our dependence on third parties in the distribution channel from our manufacturers to patients subject us to risk that limit our profitability and could limit our ability to supply Vascepa to large market segments.
We sell Vascepa principally to a limited number of major wholesalers, as well as selected regional wholesalers and specialty pharmacy providers, or collectively, our Distributors or our customers, that in turn resell Vascepa to retail pharmacies for subsequent resale to patients and healthcare providers. These parties exercise a substantial amount of bargaining power over us given their control over large segments of the market for Vascepa. This bargaining power has led us to bear increasingly higher discounts in the sale of Vascepa. In addition, payors have broad latitude to change individual products’ formulary position or to implement other barriers that inhibit patients from receiving therapies prescribed by their healthcare professionals. These payor barriers include requirements that patients try another drug before Vascepa, known as step edits, and the requirement that prior authorization be obtained by a healthcare provider after a prescription is written before a patient will be reimbursed by their health plan for the cost of a Vascepa prescription. Further, pharmacy benefit managers implement plans that act as disincentives for Vascepa use, such as increasingly higher
49
deductibles. One practical impact of higher deductibles is that they cause patients to delay filling prescriptions for asymptomatic, chronic care medications such as hypertriglyceridemia earlier in the year, until patients meet their deductible and the cost of Vascepa is then borne more by their insurance carrier. Collectively, these dynamics negatively affect our profitability for the sale of Vascepa and could increase over time further impacting our operating results. Consolidation among these industry participants could increase the pressure from these market dynamics.
Our relationships with healthcare providers and physicians and third-party payors are subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose use to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors in the United States and elsewhere play a primary role in the recommendation and prescription of pharmaceutical products. Arrangements with third-party payors and customers can expose pharmaceutical manufacturers to broadly applicable fraud and abuse and other healthcare laws and regulations, which may constrain the business or financial arrangements and relationships through which such companies sell, market and distribute pharmaceutical products. In particular, the promotion, sales and marketing of healthcare items and services, as well as a wide range of pricing, discounting, marketing and promotion, structuring and commission(s), certain customer incentive programs and other business arrangements, are subject to extensive laws designed to prevent fraud, kickbacks, self-dealing and other abusive practices. The applicable federal and state healthcare laws and regulations laws that may affect our ability to operate include, but are not limited to:
|
|
• |
the federal Anti-Kickback Statute, which prohibits, among other things, knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for, either the referral of an individual, or the purchase, lease, order or recommendation of any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. Liability may be established without a person or entity having actual knowledge of the federal anti-kickback statute or specific intent to violate it. In addition, the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases, or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection from anti-kickback liability. Moreover, there are no safe harbors for many common practices, such as educational and research grants or patient or product support programs; |
|
|
• |
the federal Civil False Claims Act (FCA), which prohibits, among other things, any person from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds, or knowingly making or using, or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly concealing, or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. The FCA also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the statute and to share in any monetary recovery. Recently, several pharmaceutical and other healthcare companies have been investigated or faced enforcement actions under the FCA for a variety of alleged improper marketing activities, including allegations that they caused false claims to be submitted because of the company’s marketing of the product for unapproved, and thus allegedly non-reimbursable, uses. Federal enforcement agencies also have showed increased interest in pharmaceutical companies’ product and patient assistance programs, including reimbursement and co-pay support services, and a number of investigations into these programs have resulted in significant civil and criminal settlements. Pharmaceutical and other healthcare companies also are subject to other federal false claims laws, including, among others, federal criminal healthcare fraud and false statement statutes that extend to non-government health benefit programs; |
|
|
• |
HIPAA, which, among other things, imposes criminal and civil liability for knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payor and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services; |
|
|
• |
HIPAA, and its implementing regulations, which impose, among other things, requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform services for them that involve the use, or disclosure of, individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions; |
50
|
|
• |
federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
|
|
• |
analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; and other state or local laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; restrict the ability of manufacturers to offer co-pay support to patients for certain prescription drugs; require drug manufacturers to report information related to clinical trials, or information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and/or require identification or licensing of sales representatives; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts |
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies continue to give regular and close scrutiny to interactions between healthcare companies and healthcare providers, and such scrutiny often leads to investigations, prosecutions, convictions and settlements in the healthcare industry. Ensuring business arrangements comply with applicable healthcare laws, as well as responding to possible investigations by government authorities, can be time- and resource-consuming and can divert a company’s attention from the business.
The failure to comply with any of these laws or regulatory requirements subjects entities to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in significant civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment, exclusion from participation in federal and state funded healthcare programs (such as Medicare and Medicaid), contractual damages and the curtailment or restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. Any action for violation of these laws, even if successfully defended, could cause a pharmaceutical manufacturer to incur significant legal expenses and divert management’s attention from the operation of the business. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, that person or entity may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. Prohibitions or restrictions on sales or withdrawal of future marketed products could materially affect business in an adverse way.
Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent inappropriate conduct may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations.
In addition, the approval and commercialization of any of our AXA Candidates outside the United States will also likely subject us to foreign equivalents of the healthcare laws mentioned above, among other foreign laws.
We rely on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including failing to meet established deadlines for the completion of such clinical trials.
Our reliance on third parties for clinical development activities reduces our control over these activities. However, if we sponsor clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trials. Moreover, the FDA requires us to comply with requirements, commonly referred to as good clinical practices, for conducting, recording, and reporting the results of clinical trials to ensure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties
51
does not relieve us of these responsibilities and requirements. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be delayed in obtaining regulatory approvals for our product candidates and may be delayed in our efforts to successfully commercialize our product candidates for targeted diseases.
Risks Related to Our Intellectual Property
We are dependent on patents, proprietary rights and confidentiality to protect the commercial potential of Vascepa.
Our success depends in part on our ability to obtain and maintain intellectual property protection for our drug candidates, technology and know-how, and to operate without infringing the proprietary rights of others. Our ability to successfully implement our business plan and to protect our products with our intellectual property will depend in large part on our ability to:
|
|
• |
obtain, defend and maintain patent protection and market exclusivity for our current and future products; |
|
|
• |
preserve any trade secrets relating to our current and future products; |
|
|
• |
acquire patented or patentable products and technologies; and |
|
|
• |
operate without infringing the proprietary rights of third parties. |
We have prosecuted, and are currently prosecuting, multiple patent applications to protect the intellectual property developed during the Vascepa development program. As of the date of this report, we had 79 patent applications in the United States that have been either issued or allowed and more than 30 additional patent applications are pending in the United States. Such 79 allowed and issued applications include the following:
|
|
• |
2 issued U.S. patents directed to a pharmaceutical composition of Vascepa in a capsule that have terms that expire in 2020 and 2030, respectively; |
|
|
• |
1 issued U.S. patent covering a composition containing highly pure EPA that expires in 2021; |
|
|
• |
43 U.S. patents covering or related to the use of Vascepa in either the MARINE or ANCHOR populations that have terms that expire in 2030 or later; |
|
|
• |
11 U.S. patents covering or related to the use of Vascepa in the REDUCE-IT population with terms expiring in 2033 or later; |
|
|
• |
4 additional patents related to the use of a pharmaceutical composition comprised of free fatty acids to treat the ANCHOR patient population with a term that expires in 2030 or later; |
|
|
• |
2 additional patents related to the use of a pharmaceutical composition comprised of free fatty acids to treat the MARINE patient population with a term that expires in 2030; |
|
|
• |
3 additional patents related to the use of a pharmaceutical composition comprised of free fatty acids to treat the REDUCE-IT population expiring 2033; |
|
|
• |
1 additional patent related to a pharmaceutical composition comprised of free fatty acids and uses thereof to treat both the MARINE and ANCHOR patient populations with a term that expires in 2030; |
|
|
• |
1 additional patent related to the use of a pharmaceutical composition comprised of re-esterified EPA triglyceride to treat the REDUCE-IT population expiring 2033; |
|
|
• |
3 additional patents related to a formulation of EPA/DHA and uses thereof with a term that expires in 2030; |
|
|
• |
2 additional patents related to the use of Vascepa to treat obesity with a term that expires in 2034; |
|
|
• |
2 additional patents covering a pharmaceutical composition comprised of EPA and a hydroxyl compound with a term that expires in 2034; and |
|
|
• |
4 additional patents covering a new combination therapy comprised of EPA and another drug. |
A Notice of Allowance is issued after the USPTO makes a determination that a patent can be granted from an application. A Notice of Allowance does not afford patent protection until the underlying patent is issued by the USPTO. No assurance can be given that applications with issued notices of allowance will be issued as patents or that any of our pending patent applications will issue as patents. No assurance can be given that, if and when issued, our patents will prevent competitors from competing with Vascepa. For example, we may choose to not assert all issued patents in patent litigation and patents or claims within patents may be determined to be invalid.
We are the owner of the above-listed patents. We are also the exclusive licensee of certain patents owned by others covering products in development. To secure our debt under our outstanding royalty-like instrument, we have granted the holders of such instrument a security interest in our Vascepa-related patents.
52
We are also pursuing patent applications related to Vascepa in multiple jurisdictions outside the United States. We may be dependent in some cases upon third-party licensors to pursue filing, prosecution and maintenance of patent rights or applications owned or controlled by those parties, including, for example, under our collaboration with Mochida. It is possible that third parties will obtain patents or other proprietary rights that might be necessary or useful to us. In cases where third parties are first to invent a particular product or technology, or first to file after various provisions of the America Invents Act of 2011 went into effect on March 16, 2013, it is possible that those parties will obtain patents that will be sufficiently broad so as to prevent us from utilizing such technology or commercializing our current and future products.
Although we intend to make reasonable efforts to protect our current and future intellectual property rights and to ensure that any proprietary technology we acquire or develop does not infringe the rights of other parties, we may not be able to ascertain the existence of all potentially conflicting claims. Therefore, there is a risk that third parties may make claims of infringement against our current or future products or technologies. In addition, third parties may be able to obtain patents that prevent the sale of our current or future products or require us to obtain a license and pay significant fees or royalties in order to continue selling such products.
We may in the future discover the existence of products that infringe patents that we own or that have been licensed to us. If we were to initiate legal proceedings against a third party to stop such an infringement, such proceedings could be costly and time consuming, regardless of the outcome. No assurances can be given that we would prevail, and it is possible that, during such a proceeding, our patent rights could be held to be invalid, unenforceable or both. Although we intend to protect our trade secrets and proprietary know-how through confidentiality agreements with our manufacturers, employees and consultants, we may not be able to prevent parties subject to such confidentiality agreements from breaching these agreements or third parties from independently developing or learning of our trade secrets.
We anticipate that competitors may from time to time oppose our efforts to obtain patent protection for new technologies or to submit patented technologies for regulatory approvals. Competitors may seek to oppose our patent applications to delay the approval process or to challenge our granted patents, for example, by requesting a reexamination of our patent at the USPTO, or by filing an opposition in a foreign patent office, even if the opposition or challenge has little or no merit. For example, one of our patents was revoked in an opposition proceeding in Europe due to a determination of improper claim amendments under a provision of law not applicable in the United States. Such proceedings are generally highly technical, expensive, and time consuming, and there can be no assurance that such a challenge would not result in the narrowing or complete revocation of any patent of ours that was so challenged.
Our issued patents may not prevent competitors from competing with Vascepa, even if we seek to enforce our patent rights.
We plan to vigorously defend our rights under issued patents. For example, in March 2014, we filed a patent infringement suit against Omthera Pharmaceuticals, Inc., and its parent company, AstraZeneca Pharmaceuticals LP. The suit sought injunctive relief and monetary damages for infringement of our U.S. Patent No. 8,663,662. The complaint alleged infringement of the patent arising from the expected launch of Epanova, a product that is expected to compete with Vascepa in the United States. The patent covers methods of lowering triglycerides by administering a pharmaceutical composition that includes amounts of EPA as free acid, and no more than about 30% DHA. In November 2014, based on a representation from AstraZeneca Pharmaceuticals LP that the commercial launch of Epanova was not imminent, the court dismissed our complaint, without prejudice (i.e., preserving our ability to later re-file the suit). The court required the defendant to notify us before any product launch. We intend to pursue this litigation vigorously and aggressively protect its intellectual property rights. However, patent litigation is a time-consuming and costly process. There can be no assurance that we will be successful in enforcing this patent or that it will not be successfully challenged and invalidated. Even if we are successful in enforcing this patent, the process could take years to reach conclusion.
Other drug companies may challenge the validity, enforceability, or both of our patents and seek to design its products around our issued patent claims and gain marketing approval for generic versions of Vascepa or branded competitive products based on new clinical studies. The pharmaceutical industry is highly competitive and many of our competitors have greater experience and resources than we have. Any such competition could undermine sales, marketing and collaboration efforts for Vascepa, and thus reduce, perhaps materially, the revenue potential for Vascepa.
Even if we are successful in enforcing our issued patents, we may incur substantial costs and divert management’s time and attention in pursuing these proceedings, which could have a material adverse effect on us. Patent litigation is costly and time consuming, and we may not have sufficient resources to bring these actions to a successful conclusion.
There can be no assurance that any of our pending patent applications relating to Vascepa or its use will issue as patents.
We have filed and are prosecuting numerous families of patent applications in the United States and internationally with claims designed to protect the proprietary position of Vascepa. For certain of these patent families, we have filed multiple patent applications. Collectively the patent applications include numerous independent claims and dependent claims. Several of our patent applications contain claims that are based upon what we believe are unexpected and favorable findings from the MARINE and ANCHOR trials. If granted, many of the resulting granted patents would expire in 2030 or beyond. However, no assurance can be given that these additional MARINE and ANCHOR patents or any of our pending patent applications intended to cover an indication based on results from the REDUCE-IT clinical trial will be granted or, if they grant, that they will prevent competitors from competing with Vascepa.
53
For example, we expect to engage in new ANDA patent litigation in the United States and elsewhere with respect to method of use patents related to the REDUCE-IT study after any newly granted indications based on that study.
Securing patent protection for a product is a complex process involving many legal and factual questions. The patent applications we have filed in the United States and internationally are at varying stages of examination, the timing of which is outside our control. The process to getting a patent granted can be lengthy and claims initially submitted are often modified in order to satisfy the requirements of the patent office. This process includes written and public communication with the patent office. The process can also include direct discussions with the patent examiner. There can be no assurance that the patent office will accept our arguments with respect to any patent application or with respect to any claim therein. The timing of the patent review process is independent of and has no effect on the timing of the FDA’s review of our NDA or sNDA submissions. We cannot predict the timing or results of any patent application. In addition, we may elect to submit, or the patent office may require, additional evidence to support certain of the claims we are pursuing. Furthermore, third parties may attempt to submit publications for consideration by the patent office during examination of our patent applications. Providing such additional evidence and publications could prolong the patent office’s review of our applications and result in us incurring additional costs. We cannot be certain what commercial value any granted patent in our patent estate will provide to us.
Despite the use of confidentiality agreements and/or proprietary rights agreements, which themselves may be of limited effectiveness, it may be difficult for us to protect our trade secrets.
In addition to our patent portfolio and strategy, we will also rely upon trade secrets and know-how to help protect our competitive position. We rely on trade secrets to protect technology in cases when we believe patent protection is not appropriate or obtainable. However, trade secrets are difficult to protect. While we require certain of our academic collaborators, contractors and consultants to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary information.
Risks Related to Our Business
If the estimates we make, or the assumptions on which we rely, in preparing our projected guidance prove inaccurate, our actual results may vary from those reflected in our projections and accruals.
In January 2019, we issued financial and business guidance, including expected fiscal year 2019 total net revenue and expectations regarding inventory build, 2019 operating expenses, and timing of an sNDA seeking Vascepa label expansion. All such guidance is based on estimates and the judgment of management. Because of the inherent nature of estimates, there could be significant differences between our estimates and the actual amount of product demand. If, for any reason, we are unable to realize our currently projected 2019 revenue, we may not realize our publicly announced financial guidance. If we fail to realize or if we change or update any element of our publicly disclosed financial guidance as we have done in the past or other expectations about our business change, our stock price could decline in value.
Potential technological changes in our field of business create considerable uncertainty.
We are engaged in the pharmaceutical field, which is characterized by extensive research efforts and rapid technological progress. New developments in research are expected to continue at a rapid pace in both industry and academia. We cannot assure you that research and discoveries by others will not render some or all of our programs or product candidates uncompetitive or obsolete. Our business strategy is based in part upon new and unproven technologies to the development of therapeutics to improve cardiovascular health. We cannot assure you that unforeseen problems will not develop with these technologies or applications or that any commercially feasible products will ultimately be developed by us.
Our internal computer systems, or those of our third‑party clinical research organizations or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our research and development programs.
Despite the implementation of security measures, our internal computer systems and those of our third‑party clinical research organizations and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. While we have not experienced any such system failure, accident, or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs. To the extent that any disruption or security breach results in a loss of or damage to our data or applications or other data or applications relating to our technology or products candidates, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities and our research and development program could be delayed.
We could be subject to risks caused by misappropriation, misuse, leakage, falsification or intentional or accidental release or loss of information maintained in the information systems and networks of our company and our vendors, including personal information of our employees and patients, and company and vendor confidential data. In addition, outside parties may attempt to penetrate our systems or those of our vendors or fraudulently induce our personnel or the personnel of our vendors to disclose sensitive information in order to gain access to our data and/or systems. We may experience threats to our data and systems, including malicious codes and viruses, phishing and other cyber-attacks. The number and complexity of these threats continue to increase over
54
time. If a material breach of our information technology systems or those of our vendors occurs, the market perception of the effectiveness of our security measures could be harmed and our reputation and credibility could be damaged. We could be required to expend significant amounts of money and other resources to repair or replace information systems or networks. In addition, we could be subject to regulatory actions and/or claims made by individuals and groups in private litigation involving privacy issues related to data collection and use practices and other data privacy laws and regulations, including claims for misuse or inappropriate disclosure of data, as well as unfair or deceptive practices. Although we develop and maintain systems and controls designed to prevent these events from occurring, and we have a process to identify and mitigate threats, the development and maintenance of these systems, controls and processes is costly and requires ongoing monitoring and updating as technologies change and efforts to overcome security measures become increasingly sophisticated. Moreover, despite our efforts, the possibility of these events occurring cannot be eliminated entirely. As we outsource more of our information systems to vendors, engage in more electronic transactions with payors and patients, and rely more on cloud-based information systems, the related security risks will increase and we will need to expend additional resources to protect our technology and information systems. In addition, there can be no assurance that our internal information technology systems or those of our third-party contractors, or our consultants’ efforts to implement adequate security and control measures, will be sufficient to protect us against breakdowns, service disruption, data deterioration or loss in the event of a system malfunction, or prevent data from being stolen or corrupted in the event of a cyberattack, security breach, industrial espionage attacks or insider threat attacks which could result in financial, legal, business or reputational harm.
We are subject to potential product liability.
Following the commercial launch of Vascepa, we will be subject to the potential risk of product liability claims relating to the manufacturing and marketing of Vascepa. Any person who is injured as a result of using Vascepa may have a product liability claim against us without having to prove that we were at fault.
In addition, we could be subject to product liability claims by persons who took part in clinical trials involving our current or former development stage products. A successful claim brought against us could have a material adverse effect on our business. We cannot guarantee that a product liability claim will not be asserted against us in the future.
A change in our tax residence could have a negative effect on our future profitability.
Under current UK legislation, a company incorporated in England and Wales, or which is centrally managed and controlled in the UK, is regarded as resident in the UK for taxation purposes. Under current Irish legislation, a company is regarded as resident for tax purposes in Ireland if it is centrally managed and controlled in Ireland, or, in certain circumstances, if it is incorporated in Ireland. Where a company is treated as tax resident under the domestic laws of both the UK and Ireland then the provisions of article 4(3) of the Double Tax Convention between the UK and Ireland provides that such enterprise shall be treated as resident only in the jurisdiction in which its place of effective management is situated. We have sought to conduct our affairs in such a way so as to be resident only in Ireland for tax purposes by virtue of having our place of effective management situated in Ireland. Trading income of an Irish company is generally taxable at the Irish corporation tax rate of 12.5%. Non-trading income of an Irish company (e.g., interest income, rental income or other passive income) is taxable at a rate of 25%.
However, we cannot assure you that we are or will continue to be resident only in Ireland for tax purposes. It is possible that in the future, whether as a result of a change in law or the practice of any relevant tax authority or as a result of any change in the conduct of our affairs, we could become, or be regarded as having become resident in a jurisdiction other than Ireland. Our and our subsidiaries’ income tax returns are periodically examined by various tax authorities, including the Internal Revenue Service (IRS) and states. We recently completed the audits by the IRS for the years 2013 to 2014, with no material changes to the filed income tax returns. Although the outcome of tax audits is always uncertain and could result in significant cash tax payments, we do not believe the outcome of any future audits will have a material adverse effect on our consolidated financial position or results of operations. The ultimate resolution may result in a payment that is materially different from our current estimate of the tax liabilities. Should we cease to be an Irish tax resident, we may be subject to a charge to Irish capital gains tax on our assets. Similarly, if the tax residency of any of our subsidiaries were to change from their current jurisdiction for any of the reasons listed above, we may be subject to a charge to local capital gains tax charge on the assets.
The effect on us of comprehensive U.S. tax reform legislation whether adverse or favorable, is uncertain.
On December 22, 2017, President Trump signed into law H.R. 1, “An Act to provide for reconciliation pursuant to titles II and V of the concurrent resolution on the budget for fiscal year 2018”, or informally, the Tax Cuts and Jobs Act. Among a number of significant changes to the U.S. federal income tax rules, the Tax Cuts and Jobs Act reduces the marginal U.S. corporate income tax rate from 35% to 21%, limits the deduction for net interest expense, shifts the United States toward a more territorial tax system, and imposes new taxes to combat erosion of the U.S. federal income tax base. The effect of the Tax Cuts and Jobs Act on our company and our affiliates, whether adverse or favorable, is uncertain, and may not become evident for some period of time. You are urged to consult your tax adviser regarding the implications of the Tax Cuts and Jobs Act for an investment in our ADSs.
55
The loss of key personnel could have an adverse effect on our business.
We are highly dependent upon the efforts of our senior management. The loss of the services of one or more members of senior management could have a material adverse effect on us. As a small company with a streamlined management structure, the departure of any key person could have a significant impact and would be potentially disruptive to our business until such time as a suitable replacement is hired. Furthermore, because of the specialized nature of our business, as our business plan progresses we will be highly dependent upon our ability to attract and retain qualified scientific, technical and key management personnel. As we evolve from a development stage company to a commercial stage company we may experience turnover among members of our senior management team. We may have difficulty identifying and integrating new executives to replace any such losses. There is intense competition for qualified personnel in the areas of our activities. In this environment, we may not be able to attract and retain the personnel necessary for the development of our business, particularly if we do not achieve profitability. The failure to recruit key scientific, technical and management personnel would be detrimental to our ability to implement our business plan.
We could be adversely affected by our exposure to customer concentration risk.
A significant portion of our sales are to wholesalers in the pharmaceutical industry. Three customers individually accounted for 10% or more of our gross product sales. Customers A, B, and C accounted for 31%, 30%, and 27%, respectively, of gross product sales for the year ended December 31, 2018 and represented 26%, 24%, and 39%, respectively, of the gross accounts receivable balance as of December 31, 2018. Customers A, B, and C accounted for 27%, 33%, and 28%, respectively, of gross product sales for the year ended December 31, 2017 and represented 27%, 21%, and 41%, respectively, of the gross accounts receivable balance as of December 31, 2017. There can be no guarantee that we will be able to sustain our accounts receivable or gross sales levels from our key customers. If, for any reason, we were to lose, or experience a decrease in the amount of business with our largest customers, whether directly or through our distributor relationships, our financial condition and results of operations could be negatively affected.
Risks Related to Our Financial Position and Capital Requirements
We have a history of operating losses and anticipate that we will incur continued losses for an indefinite period of time.
We have not yet reached profitability. For the fiscal years ended December 31, 2018, 2017, and 2016, we reported losses of approximately $116.4 million, $67.9 million, and $86.4 million, respectively, and we had an accumulated deficit as of December 31, 2018 of $1.4 billion. Substantially all of our operating losses resulted from costs incurred in connection with our research and development programs, from general and administrative costs associated with our operations, and costs related to the commercialization of Vascepa. Additionally, as a result of our significant expenses relating to research and development and to commercialization, we expect to continue to incur significant operating losses for an indefinite period. Because of the numerous risks and uncertainties associated with developing and commercializing pharmaceutical products, we are unable to predict the magnitude of these future losses. Our historic losses, combined with expected future losses, have had and will continue to have an adverse effect on our cash resources, shareholders’ deficit and working capital.
Although we began generating revenue from Vascepa in January 2013, we may never be profitable.
Our ability to become profitable depends upon our ability to generate revenue. We have been generating product revenue from sales of Vascepa since January 2013, but we may not be able to generate sufficient revenue to attain profitability. Our ability to generate profits on sales of Vascepa is subject to the market acceptance and commercial success of Vascepa and our ability to manufacture commercial quantities of Vascepa through third parties at acceptable cost levels, and may also depend upon our ability to effectively market and sell Vascepa through our strategic collaborations.
Even though Vascepa has been approved by the FDA for marketing in the United States in the MARINE indication, it may not gain market acceptance or achieve commercial success and it may never be approved for the ANCHOR indication or any other indication. In addition, we anticipate continuing to incur significant costs associated with commercializing Vascepa. We may not achieve profitability in the near term due to high costs associated with our REDUCE-IT study and commercialization efforts, for example. If we are unable to continue to generate robust product revenues, we will not become profitable in the near term, if ever, and may be unable to continue operations without continued funding.
Our historical financial results do not form an accurate basis for assessing our current business.
As a consequence of the many years developing Vascepa for commercialization and the commercial launch of Vascepa in 2013 in the United States, our historical financial results do not form an accurate basis upon which investors should base their assessment of our business and prospects. In addition, we expect that our costs will increase substantially as we continue to commercialize Vascepa in the MARINE indication and with ANCHOR data and seek to obtain additional regulatory approval of Vascepa from the REDUCE-IT cardiovascular outcomes study. Accordingly, our historical financial results reflect a substantially different business from that currently being conducted and from that expected in the future. In addition, we have a limited history of obtaining regulatory approval for, and no demonstrated ability to successfully commercialize, a product candidate. Consequently, any predictions about our future
56
performance may not be as accurate as they could be if we had a history of successfully developing and commercializing pharmaceutical products.
Our operating results are unpredictable and may fluctuate. If our operating results are below the expectations of securities analysts or investors, the trading price of our stock could decline.
Our operating results are difficult to predict and will likely fluctuate from quarter to quarter and year to year, and Vascepa prescription figures will likely fluctuate from month to month. Vascepa sales are difficult to predict from period to period and as a result, you should not rely on Vascepa sales results in any period as being indicative of future performance, and sales of Vascepa may be below the expectation of securities analysts or investors in the future. We believe that our quarterly and annual results of operations may be affected by a variety of factors, including:
|
|
• |
the continuing evolution of the medical community’s and the public’s perception of the REDUCE-IT study results; |
|
|
• |
the level of demand for Vascepa, due to changes in prescriber sentiment, quarterly changes in Distributor purchases, and other factors; |
|
|
• |
the extent to which coverage and reimbursement for Vascepa is available from government and health administration authorities, private health insurers, managed care programs and other third-party payers and the timing and extent to which such coverage and reimbursement changes; |
|
|
• |
the timing, cost and level of investment in our sales and marketing efforts to support Vascepa sales and the resulting effectiveness of those efforts with our co-promotion partner, Kowa Pharmaceuticals America, Inc.; |
|
|
• |
the timing and ability of commercialization partners outside the United States, to develop, register and commercialize Vascepa in the China Territory, several Middle Eastern and North African countries, and Canada, for example, including obtaining necessary regulatory approvals and establishing marketing channels; |
|
|
• |
additional developments regarding our intellectual property portfolio and regulatory exclusivity protections, if any; |
|
|
• |
outcomes of litigation and other legal proceedings; and |
|
|
• |
our regulatory dialogue on the REDUCE-IT study. |
We may require substantial additional resources to fund our operations. If we cannot find additional capital resources, we will have difficulty in operating as a going concern and growing our business.
We currently operate with limited resources. We believe that our cash and cash equivalents balance of $249.2 million as of December 31, 2018, will be sufficient to fund our projected operations for at least twelve months and through the likely Prescription Drug User Fee Act (PDUFA) date for approval of a supplemental new drug application (sNDA) by the FDA based on REDUCE-IT study results. Depending on the level of cash generated from operations, and depending in part on the timing and results of the FDA review of the sNDA and rate of prescription growth for Vascepa, additional capital may be required to support planned expansion of Vascepa promotion and potential Vascepa promotion beyond which we are currently executing. If additional capital is required and we are unable to obtain additional capital, we may be forced to delay, limit or eliminate certain promotional activities. We anticipate that quarterly net cash outflows in future periods will be variable.
In order to fully realize the market potential of Vascepa, we may need to enter into a new strategic collaboration or raise additional capital.
Our future capital requirements will depend on many factors, including:
|
|
• |
the timing, amount and consistency of revenue generated from the commercial sale of Vascepa; |
|
|
• |
the costs associated with commercializing Vascepa in the United States, including expenditures such as potential direct-to-consumer advertising and increased sales force sizing, and for additional indications in the United States and in jurisdictions in which we receive regulatory approval, if any, the cost and timing of securing commercial supply of Vascepa and the timing of entering into any new strategic collaboration with others relating to the commercialization of Vascepa, if at all, and the terms of any such collaboration; |
|
|
• |
continued costs associated with litigation and other legal proceedings; |
|
|
• |
the time and costs involved in obtaining additional regulatory approvals for Vascepa based on REDUCE-IT results; |
|
|
• |
the extent to which we continue to develop internally, acquire or in-license new products, technologies or businesses; and |
|
|
• |
the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights. |
If we require additional funds and adequate funds are not available to us in amounts or on terms acceptable to us or on a timely basis, or at all, our commercialization efforts for Vascepa may suffer materially.
57
The potential future benefit of our substantial net operating loss carryforwards could be lost and our prospects for profitability could be materially diminished if tax regulations or rates change or if we are deemed to not have active operations in Ireland.
Tax law and policies in the United States and Ireland are subject to change based on adjustments in political perspectives. In the United States and internationally, how to tax entities with international operations, like Amarin, has been subject to significant re-evaluation. We developed Vascepa in and from Ireland. In recent years, particularly since 2013 when commercial sale of Vascepa commenced in the United States, the majority of our consolidated operations have been in the United States. Ownership to Vascepa continues to reside with our wholly-owned Ireland-based subsidiary, Amarin Pharmaceuticals Ireland Ltd., and oversight and operations of that entity are structured to be maintained in Ireland. In order to effectively utilize our accumulated net operating loss carryforwards for tax purposes in Ireland, our operations, particularly for this subsidiary, need to be active in Ireland. In addition, utilization of these accumulated net operating loss carryforwards assumes that tax treaties between Ireland and other countries, particularly the United States, do not change in a manner which limit our future ability to offset earnings with these operating loss carryforwards for tax purposes.
Similarly, a change in our Irish tax residence could materially affect our ability to obtain profitability, if at all. Changes in tax law and tax rates, particularly in the United States and Ireland, could also impact our assessment of deferred taxes. Any change in our assessment of the realizability or the timing for realizing deferred taxes could have a negative impact our future profitability.
Continued negative economic conditions would likely have a negative effect on our ability to obtain financing on acceptable terms.
While we may seek additional funding through public or private financings, we may not be able to obtain financing on acceptable terms, or at all. There can be no assurance that we will be able to access equity or credit markets in order to finance our current operations or expand development programs for Vascepa, or that there will not be a further deterioration in financial markets and confidence in economies. We may also have to scale back or further restructure our operations. If we are unable to obtain additional funding on a timely basis, we may be required to curtail or terminate some or all of our research or development programs or our commercialization strategies.
Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights.
To the extent we are permitted under our December 2012 Purchase and Sale Agreement with CPPIB Credit Europe S.à r.l., or CPPIB, as successor in interest to BioPharma Secured Debt Fund II Holdings Cayman LP, we may seek additional capital through a combination of private and public equity offerings, debt financings and collaboration, strategic and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a shareholder.
Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration, strategic alliance and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, Vascepa or product candidates beyond the rights we have already relinquished, or grant licenses on terms that are not favorable to us.
Potential business combinations or other strategic transactions may disrupt our business or divert management’s attention.
On a regular basis, we explore potential business combination transactions, including an acquisition of us by a third party, exclusive licenses of Vascepa or other strategic transactions or collaborations with third parties. For example, in March 2014, we entered into a co-promotion agreement with Kowa Pharmaceuticals America, Inc. related to the commercialization of Vascepa in the United States. The consummation and performance of any such future transactions or collaborations will involve risks, such as:
|
|
• |
diversion of managerial resources from day-to-day operations; |
|
|
• |
exposure to litigation from the counterparties to any such transaction, other third parties or our shareholders; |
|
|
• |
misjudgment with respect to the value; |
|
|
• |
higher than expected transaction costs; or |
|
|
• |
an inability to successfully consummate any such transaction or collaboration. |
As a result of these risks, we may not be able to achieve the expected benefits of any such transaction or collaboration or deliver the value thereof to our shareholders. If we are unsuccessful in consummating any such transaction or collaboration, we may be required to reevaluate our business only after we have incurred substantial expenses and devoted significant management time and resources.
58
Risks Related to Ownership of our ADSs and Common Shares
The price of our ADSs and common shares may be volatile.
The stock market has from time to time experienced significant price and volume fluctuations that may be unrelated to the operating performance of particular companies. In addition, the market prices of the securities of many pharmaceutical and medical technology companies have been especially volatile in the past, and this trend is expected to continue in the future.
As of February 22, 2019, we had 329,321,488 common shares outstanding including 329,087,415 shares held as ADSs and 234,073 held as ordinary shares (which are not held in the form of ADSs). There is a risk that there may not be sufficient liquidity in the market to accommodate significant increases in selling activity or the sale of a large block of our securities. Our ADSs have historically had limited trading volume, which may also result in volatility. If any of our large investors seek to sell substantial amounts of our ADSs, particularly if these sales are in a rapid or disorderly manner, or other investors perceive that these sales could occur, the market price of our ADSs could decrease significantly.
The market price of our ADSs and common shares may also be affected by factors such as:
|
|
• |
developments or disputes concerning ongoing patent prosecution efforts and any future patent or proprietary rights; |
|
|
• |
litigation and regulatory developments in the United States affecting our Vascepa promotional rights, and regulatory developments in other countries; |
|
|
• |
actual or potential medical results relating to our products or our competitors’ products; |
|
|
• |
interim failures or setbacks in product development; |
|
|
• |
innovation by us or our competitors; |
|
|
• |
currency exchange rate fluctuations; and |
|
|
• |
period-to-period variations in our results of operations. |
The number of our ordinary shares, or ADSs representing such ordinary shares, outstanding may increase substantially as a result of our March 2015 private placement and the later consolidation and redesignation of the Series A Preference Shares represented by Preference ADSs issued thereunder, and some of the investors may then beneficially own significant blocks of our ordinary shares; the ordinary shares and Series A Preference Shares resulting from the private placement will be generally available for resale in the public market upon registration under the Securities Act.
In March and July 2015, we completed a private placement of American Depositary Shares in two tranches representing 352,150,790 and 38,867,180 Series A Preference Shares, respectively, each ten (10) of which may be consolidated and redesignated into one (1) ordinary share in our capital. During the three months ended June 30, 2015, 62,833,330 preferred shares were converted, resulting in the issuance of 6,283,333 ordinary shares and during the three months ended September 30, 2018, 38,867,180 preferred shares were converted, resulting in the issuance of 3,886,718 ordinary shares. The consolidation and redesignation of the Series A Preference Shares currently outstanding would result in an additional 28,931,746 ordinary shares outstanding, resulting in substantial dilution to shareholders who held our ordinary shares or ADSs representing such ordinary shares prior to the private placement. Although the Series A Preference Shares do not have voting rights, in general, upon consolidation and redesignation into ordinary shares some of the investors in the private placement could then have significant influence over the outcome of any shareholder vote, including the election of directors and the approval of mergers or other business combination transactions.
Pursuant to the securities subscription agreements that we entered into with the investors in the private placement, we agreed to file with the SEC a registration statement to register the resale of the Series A Preference Shares represented by American Depositary Shares issued in the private placement and the ordinary shares issuable upon the consolidation and consolidation and redesignation of such Series A Preference Shares. Upon such registration and subsequent consolidation and redesignation, these securities will become generally available for immediate resale in the public market. The market price of our ordinary shares could fall as a result of an increase in the number of shares available for sale in the public market.
Failure to comply with our obligations under the March 2015 securities subscription agreements could result in our becoming liable for damages to certain investors under these agreements, including specified liquidated damages, which could be material in amount.
Under the terms of the March 2015 securities subscription agreements, we are subject to various obligations, failure to comply with which could result in our becoming liable to certain investors under these agreement for damages, which could be material in amount.
For example, under each of these agreements we have agreed to file and maintain the effectiveness of certain resale registration statements for ADSs representing the ordinary shares underlying the Series A Preference shares we issued and sold under these
59
agreements. Specifically, we have agreed to pay liquidated damages to the investors in the respective private placements if (a) the applicable resale registration statements we are required to file are not declared effective within 120 days after the closing of the applicable private placement, or (b) after effectiveness and subject to certain specified exceptions, we suspend the use of the applicable registration statement or the registration statement ceases to remain continuously effective as to all the securities for which it is required to be effective. We refer to each of these events as a registration default. Subject to the specified exceptions, for each 30-day period or portion thereof during which a registration default remains uncured, we are obligated to pay liquidated damages to each investor in cash in an amount equal to 1% of the aggregate subscription price paid by each such investor in the private placement, up to a maximum of 8% of such aggregate subscription price. These amounts could be material, and any liquidated damages we are required to pay could have a material adverse effect on our financial condition.
In addition, under the securities subscription agreement dated as of March 5, 2015, we are required to not publicly disclose the identity of the investors party to that agreement, subject to certain exceptions for disclosures required in securities filings and under applicable law. If we fail to comply with these obligations we could become liable to these investors for damages, including specified liquidated damages. For example, following certain public statements made by us on a quarterly conference call concerning the 2015 private placement, we agreed to specified liquidated damages in the event we are found to have violated the confidentiality provisions of the subscription agreement in the future.
Actual or potential sales of our common shares by our employees, including members of our senior management team, pursuant to pre-arranged stock trading plans could cause our stock price to fall or prevent it from increasing for numerous reasons, and actual or potential sales by such persons could be viewed negatively by other investors.
In accordance with the guidelines specified under Rule 10b5-1 of the Securities Exchange Act of 1934 and our policies regarding stock transactions, a number of our directors and employees, including members of our senior management team, have adopted and may continue to adopt pre-arranged stock trading plans to sell a portion of our common stock. Generally, sales under such plans by members of our senior management team and directors require public filings. Actual or potential sales of our ADSs by such persons could cause the price of our ADSs to fall or prevent it from increasing for numerous reasons. For example, a substantial amount of our ADSs becoming available (or being perceived to become available) for sale in the public market could cause the market price of our ADSs to fall or prevent it from increasing. Also, actual or potential sales by such persons could be viewed negatively by other investors.
We may be a passive foreign investment company, or PFIC, which would result in adverse U.S. federal tax consequences to U.S. investors.
Amarin Corporation plc and certain of our subsidiaries may be classified as “passive foreign investment companies,” or PFICs, for U.S. federal income tax purposes. The tests for determining PFIC status for a taxable year depend upon the relative values of certain categories of assets and the relative amounts of certain kinds of income. The application of these factors depends upon our financial results, which are beyond our ability to predict or control, and which may be subject to legal and factual uncertainties.
We believe it is prudent to assume that we were classified as a PFIC in the past. However, we do not believe that we have been classified as a PFIC beginning in 2013 when we commercially launched Vascepa in the United States and began to derive revenues from sales of Vascepa. Our status as a PFIC is subject to change in future years.
If we are a PFIC, U.S. holders of notes, ordinary shares or ADSs would be subject to adverse U.S. federal income tax consequences, such as ineligibility for any preferred tax rates on capital gains or on actual or deemed dividends, interest charges on certain taxes treated as deferred, and additional reporting requirements under U.S. federal income tax laws and regulations. Whether or not U.S. holders of our ADSs make a timely “QEF election” or “mark-to-market election” may affect the U.S. federal income tax consequences to U.S. holders with respect to the acquisition, ownership and disposition of Amarin ADSs and any distributions such U.S. holders may receive. A QEF election and other elections that may mitigate the effect of our being classified as a PFIC are unavailable with respect to the notes. Investors should consult their own tax advisors regarding all aspects of the application of the PFIC rules to the notes, ordinary shares and ADSs.
Failure to meet our obligations under our December 2012 Purchase and Sale Agreement could adversely affect our financial results and liquidity.
Pursuant to our December 2012 Purchase and Sale Agreement with CPPIB, which was assigned to CPPIB by BioPharma Secured Debt Fund II Holdings Cayman LP in December 2017, we are obligated to make payments based on the amount of our net product sales of Vascepa and any future products based on ethyl-EPA, or covered products, subject to certain quarterly caps.
Pursuant to this agreement, we may not, among other things: (i) incur indebtedness greater than a specified amount, which we refer to as the Indebtedness Covenant; (ii) pay a dividend or other cash distribution, unless we have cash and cash equivalents in excess of a specified amount after such payment; (iii) amend or restate our memorandum and articles of association unless such
60
amendments or restatements do not affect CPPIB’s interests under the transaction; (iv) encumber any of the collateral securing our performance under the agreement; and (v) abandon certain patent rights, in each case without the consent of CPPIB.
Upon a transaction resulting in a change of control of Amarin, as defined in the agreement, CPPIB will be automatically entitled to receive any amounts not previously paid, up to our maximum repayment obligation. As defined in the agreement, “change of control” includes, among other things, (i) a greater than 50 percent change in the ownership of Amarin, (ii) a sale or disposition of any collateral securing our debt with CPPIB and (iii), unless CPPIB has been paid a certain amount under the indebtedness, certain licensings of Vascepa to a third party for sale in the United States. The acceleration of the payment obligation in the event of a change of control transaction may make us less attractive to potential acquirers, and the payment of such funds out of our available cash or acquisition proceeds would reduce acquisition proceeds for our shareholders.
To secure our obligations under the agreement, we granted CPPIB a security interest in our rights in patents, trademarks, trade names, domain names, copyrights, know-how and regulatory approvals related to the covered products, all books and records relating to the foregoing and all proceeds of the foregoing, which we refer to as the collateral. If we (i) fail to deliver a payment when due and do not remedy that failure within specific notice period, (ii) fail to maintain a first-priority perfected security interest in the collateral in the United States and do not remedy that failure after receiving notice of such failure or (iii) become subject to an event of bankruptcy, then CPPIB may attempt to collect the maximum amount payable by us under this agreement (after deducting any payments we have already made).
There can be no assurance that we will not breach the covenants or other terms of, or that an event of default will not occur under, this agreement and, if a breach or event of default occurs, there can be no assurance that we will be able to cure the breach within the time permitted. Any failure to pay our obligations when due, any breach or default of our covenants or other obligations, or any other event that causes an acceleration of payment at a time when we do not have sufficient resources to meet these obligations, could have a material adverse effect on our business, results of operations, financial condition and future viability.
Our indebtedness could adversely affect our financial condition.
Our indebtedness and the related annual debt service requirements may adversely impact our business, operations and financial condition in the future. For example, they could:
|
|
• |
increase our vulnerability to general adverse economic and industry conditions; |
|
|
• |
limit our ability to raise additional funds by borrowing or engaging in equity sales in order to fund future working capital, capital expenditures, research and development and other general corporate requirements; |
|
|
• |
require us to dedicate a substantial portion of our cash to service payments on our debt or to restructure our debt; or |
|
|
• |
limit our flexibility to react to changes in our business and the industry in which we operate or to pursue certain strategic opportunities that may present themselves. |
We do not intend to pay cash dividends on the ordinary shares in the foreseeable future.
We have never paid dividends on ordinary shares and do not anticipate paying any cash dividends on the ordinary shares in the foreseeable future. Under English law, any payment of dividends would be subject to relevant legislation and our Articles of Association, which requires that all dividends must be approved by our Board of Directors and, in some cases, our shareholders, and may only be paid from our distributable profits available for the purpose, determined on an unconsolidated basis.
The rights of our shareholders may differ from the rights typically offered to shareholders of a U.S. corporation.
We are incorporated under English law. The rights of holders of ordinary shares and, therefore, certain of the rights of holders of ADSs, are governed by English law, including the provisions of the Companies Act 2006, and by our Articles of Association. These rights differ in certain respects from the rights of shareholders in typical U.S. corporations. The principal differences include the following:
|
|
• |
Under English law and our Articles of Association, each shareholder present at a meeting has only one vote unless demand is made for a vote on a poll, in which case each holder gets one vote per share owned. Under U.S. law, each shareholder typically is entitled to one vote per share at all meetings. |
|
|
• |
Under English law, it is only on a poll that the number of shares determines the number of votes a holder may cast. You should be aware, however, that the voting rights of ADSs are also governed by the provisions of a deposit agreement with our depositary bank. |
61
|
|
into, ordinary shares for cash. Under U.S. law, shareholders generally do not have preemptive rights unless specifically granted in the certificate of incorporation or otherwise. |
|
|
• |
Under English law and our Articles of Association, certain matters require the approval of 75% of the shareholders who vote (in person or by proxy) on the relevant resolution (or on a poll of shareholders representing 75% of the ordinary shares voting (in person or by proxy)), including amendments to the Articles of Association. This may make it more difficult for us to complete corporate transactions deemed advisable by our board of directors. Under U.S. law, generally only majority shareholder approval is required to amend the certificate of incorporation or to approve other significant transactions. |
|
|
• |
In the United Kingdom, takeovers may be structured as takeover offers or as schemes of arrangement. Under English law, a bidder seeking to acquire us by means of a takeover offer would need to make an offer for all of our outstanding ordinary shares/ADSs. If acceptances are not received for 90% or more of the ordinary shares/ADSs under the offer, under English law, the bidder cannot complete a “squeeze out” to obtain 100% control of us. Accordingly, acceptances of 90% of our outstanding ordinary shares/ADSs will likely be a condition in any takeover offer to acquire us, not 50% as is more common in tender offers for corporations organized under Delaware law. By contrast, a scheme of arrangement, the successful completion of which would result in a bidder obtaining 100% control of us, requires the approval of a majority of shareholders voting at the meeting and representing 75% of the ordinary shares voting for approval. |
|
|
• |
Under English law and our Articles of Association, shareholders and other persons whom we know or have reasonable cause to believe are, or have been, interested in our shares may be required to disclose information regarding their interests in our shares upon our request, and the failure to provide the required information could result in the loss or restriction of rights attaching to the shares, including prohibitions on certain transfers of the shares, withholding of dividends and loss of voting rights. Comparable provisions generally do not exist under U.S. law. |
|
|
• |
The quorum requirement for a shareholders’ meeting is a minimum of two shareholders entitled to vote at the meeting and present in person or by proxy or, in the case of a shareholder which is a corporation, represented by a duly authorized officer. Under U.S. law, a majority of the shares eligible to vote must generally be present (in person or by proxy) at a shareholders’ meeting in order to constitute a quorum. The minimum number of shares required for a quorum can be reduced pursuant to a provision in a company’s certificate of incorporation or bylaws, but typically not below one-third of the shares entitled to vote at the meeting. |
Shareholder protections found in provisions under the U.K. City Code on Takeovers and Mergers, or the Takeover Code, do not apply to us.
The Takeover Code provides a framework within which takeovers of certain companies organized in the United Kingdom are regulated and conducted. However, because our place of central management and control is currently outside of the United Kingdom, we are not subject to the Takeover Code. As a result, our shareholders are not entitled to the benefit of certain takeover offer protections provided under the Takeover Code. The following is a brief summary of some of the most important rules of the Takeover Code which, as noted, does not apply to us:
|
|
• |
In connection with a potential offer, if following an approach by or on behalf of a potential bidder, the company is “the subject of rumor or speculation” or there is an “untoward movement” in the company’s share price, there is a requirement for the potential bidder to make a public announcement about a potential offer for the company, or for the company to make a public announcement about its review of a potential offer. |
|
|
• |
When a person or group (a) acquires interests in shares carrying 30% or more of the voting rights of a company (which percentage is treated by the Takeover Code as the level at which effective control is obtained) or (b) increases the aggregate percentage interest they have when they are already interested in not less than 30% and not more than 50%, they must make a cash offer to all other shareholders at the highest price paid by them in the 12 months before the offer was announced. |
|
|
• |
When interests in shares carrying 10% or more of the voting rights of a class have been acquired by an offeror (i.e., a bidder) in the offer period (i.e., before the shares subject to the offer have been acquired) and the previous 12 months, the offer must be in cash or be accompanied by a cash alternative for all shareholders of that class at the highest price paid by the offeror in that period. Further, if an offeror acquires any interest in shares during the offer period, the offer for the shares must be in cash or accompanied by a cash alternative at a price at least equal to the price paid for such shares during the offer period. |
|
|
• |
If after an announcement is made, the offeror acquires an interest in shares in an offeree company (i.e., a target) at a price higher than the value of the offer, the offer must be increased accordingly. |
|
|
• |
The offeree company must appoint a competent independent adviser whose advice on the financial terms of the offer must be made known to all the shareholders, together with the opinion of the board of directors of the offeree company. |
62
|
|
• |
All shareholders must be given the same information. |
|
|
• |
Those issuing takeover circulars must include statements taking responsibility for the contents thereof. |
|
|
• |
Profit forecasts, quantified financial benefits statements and asset valuations must be made to specified standards and must be reported on by professional advisers. |
|
|
• |
Misleading, inaccurate or unsubstantiated statements made in documents or to the media must be publicly corrected immediately. |
|
|
• |
Actions during the course of an offer by the offeree company, which might frustrate the offer are generally prohibited unless shareholders approve these plans. Frustrating actions would include, for example, lengthening the notice period for directors under their service contract or agreeing to sell off material parts of the target group. |
|
|
• |
Stringent requirements are laid down for the disclosure of dealings in relevant securities during an offer, including the prompt disclosure of positions and dealing in relevant securities by the parties to an offer and any person who is interested (directly or indirectly) in 1% or more of any class of relevant securities. |
|
|
• |
Employees of both the offeror and the offeree company and the trustees of the offeree company’s pension scheme must be informed about an offer. In addition, the offeree company’s employee representatives and pension scheme trustees have the right to have a separate opinion on the effects of the offer on employment appended to the offeree board of directors’ circular or published on a website. |
U.S. shareholders may not be able to enforce civil liabilities against us.
We are incorporated under the laws of England and Wales, and our subsidiaries are incorporated in various jurisdictions, including foreign jurisdictions. A number of the officers and directors of each of our subsidiaries are non-residents of the United States, and all or a substantial portion of the assets of such persons are located outside the United States. As a result, it may not be possible for investors to affect service of process within the United States upon such persons or to enforce against them judgments obtained in U.S. courts predicated upon the civil liability provisions of the federal securities laws of the United States. We have been advised by our English solicitors that there is doubt as to the enforceability in England in original actions, or in actions for enforcement of judgments of U.S. courts, of civil liabilities to the extent predicated upon the federal securities laws of the United States.
U.S. holders of the ADSs or ordinary shares may be subject to U.S. federal income taxation at ordinary income tax rates on undistributed earnings and profits.
There is a risk that we will be classified as a controlled foreign corporation, or CFC, for U.S. federal income tax purposes. If we are classified as a CFC, any ADS holder or shareholder that is a U.S. person that owns directly, indirectly or by attribution, 10% or more of the voting power of our outstanding shares may be subject to U.S. income taxation at ordinary income tax rates on all or a portion of our undistributed earnings and profits attributable to “subpart F income.” Such 10% holder may also be taxable at ordinary income tax rates on any gain realized on a sale of ordinary shares or ADS, to the extent of our current and accumulated earnings and profits attributable to such shares. The CFC rules are complex and U.S. holders of the ordinary shares or ADSs are urged to consult their own tax advisors regarding the possible application of the CFC rules to them in their particular circumstances.
None.
The following table lists the location, use and ownership interest of our principal properties as of February 22, 2019:
|
Location |
|
Use |
|
Ownership |
|
Size (sq. ft.) |
|
||
|
Dublin, Ireland |
|
|
Offices |
|
Leased |
|
|
270 |
|
|
Bedminster, New Jersey, USA |
|
|
Offices |
|
Leased |
|
|
27,951 |
|
Effective November 1, 2011, we leased 320 square feet of office space in Dublin, Ireland. The office space was subsequently reduced to 270 square feet, effective November 1, 2013. The lease terminates on October 31, 2019 and may be renewed annually.
63
Effective July 1, 2011, we leased 9,747 square feet of office space in Bedminster, New Jersey. On December 6, 2011 we leased an additional 2,142 square feet of space in the same location. On December 15, 2012 and May 8, 2013, we leased an additional 2,601 and 10,883 square feet of space, respectively, in the same location. In January 2014 and April 2014, we entered into separate transactions with the landlord of this property to vacate approximately 2,142 and 2,000 square feet of space in exchange for discounts on contractual future rent payments. In January 2015, we signed an agreement to sublease approximately 4,700 square feet of this property to a third party, effective April 1, 2015. This sublease agreement was terminated as of September 30, 2017. Additionally, in June 2015, we executed an agreement to sublease approximately 2,500 square feet of this property to a separate third party, effective June 16, 2015, which agreement naturally ceased on March 31, 2018. On December 15, 2016, we leased an additional 732 square feet of space in the same location, effective January 1, 2017. The lease, as amended, terminates on April 30, 2019, but we are currently in the process of extending this lease through the start of the new Bridgewater, New Jersey lease, as described below. On January 26, 2019, we leased an additional 5,988 square feet in an annex building, effective February 1, 2019 and terminating June 30, 2019.
As described more fully in Note 17—Subsequent Events, given the anticipated expiration in 2019 of the lease described above, on February 5, 2019, we entered into a lease agreement for approximately 67,747 square feet of office space in Bridgewater, New Jersey with a commencement date anticipated to be on or about July 1, 2019, which will better suit our needs going forward. We believe that our existing facilities and disclosed plans for new facilities are adequate for our current needs and that additional space will be available in the future on commercially reasonable terms as needed.
On February 22, 2019, a purported investor in our publicly traded securities filed a putative class action lawsuit against Amarin Corporation plc, our chief executive officer and chief scientific officer in the U.S. District Court for the District of New Jersey, Debendra Sharma v. Amarin Corporation plc, John F. Thero and Steven Ketchum, No. 2:19-cv-06601 (D.N.J. Feb. 22, 2019). The lawsuit alleges that, during the period September 24, 2018 and November 9, 2018, we misled investors by purportedly not disclosing that the placebo given to patients in the REDUCE-IT study, mineral oil, may have caused cardiovascular problems in the patients taking it, thereby misleading investors on the outcome of the REDUCE-IT study and artificially inflating the price of our securities. Based on these allegations, the suit asserts claims under the Securities Exchange Act of 1934 and seeks unspecified monetary damages and attorneys’ fees and costs. We believe that we have valid defenses and we will vigorously defend against the claims, but cannot predict the outcome of this lawsuit. We are unable to reasonably estimate the loss exposure, if any, associated with the claims. We have insurance coverage that is anticipated to cover any significant loss exposure that may arise from this action after payment by us of the associated deductible obligation under such insurance coverage.
On August 30, 2017, Amarin Pharma, Inc. and Amarin Pharmaceuticals Ireland Limited, each wholly-owned subsidiaries of Amarin Corporation plc, filed a lawsuit with the United States International Trade Commission, or the ITC, captioned In the Matter of Certain Synthetically Produced, Predominantly EPA Omega-3 Products in Ethyl Ester or Re-esterified Triglyceride Form, USITC Docket 337-3247, against manufacturers, importers, and distributors of products containing synthetically produced omega-3 products in ethyl ester or re-esterified triglyceride form that contain more EPA than DHA or any other single component for use in or as dietary supplements. The lawsuit sought an investigation by the ITC under Section 337 of the Tariff Act of 1930 (19 U.S.C. §1337), which makes unlawful unfair methods of competition and unfair acts involving the importation and sale of articles in the United States that injure or threaten injury to a domestic industry. On October 27, 2017, the ITC determined to not institute our requested investigation. On December 1, 2017, we appealed the ITC’s non-institution decision to the United States Court of Appeals for the Federal Circuit (Case Nos. 18-1247, 18-114). That appeal is ongoing. We intend to pursue this matter vigorously.
In September and October 2016, we received paragraph IV certification notices from four companies contending to varying degrees that certain of our patents are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of Vascepa as described in those companies’ abbreviated new drug applications, or ANDAs. We filed patent infringement lawsuits against three of these four ANDA applicants. In October 2016, Amarin filed a lawsuit against Roxane Laboratories, Inc. and related parties (collectively, “Roxane”) in the U.S. District Court for the District of Nevada. The case against Roxane is captioned Amarin Pharma, Inc. et al. v. Roxane Laboratories, Inc. et al., Civ. A. No. 2:16-cv-02525 (D. Nev.). According to a stipulation filed with the Nevada court, in December 2016, Roxane transferred its ANDA to West-Ward Pharmaceuticals International Limited, which then designated West-Ward Pharmaceuticals Corp. (or together with West-Ward Pharmaceuticals International Limited, West-Ward) as its agent for FDA communications. In view of the ANDA transfer, in February 2017, West-Ward replaced Roxane and related parties as Defendants in the above-referenced case. The case against West-Ward is now captioned Amarin Pharma, Inc. et al. v. West-Ward Pharmaceuticals Corp. et al., Civ. A. No. 2:16-cv-02525 (D. Nev.). In November 2016, Amarin filed a lawsuit against Dr. Reddy’s Laboratories, Inc. and Dr. Reddy’s Laboratories, Ltd. (collectively, “DRL”) in the U.S. District Court for the District of Nevada. The case against DRL is captioned Amarin Pharma, Inc. et al. v. Dr. Reddy’s Laboratories, Inc. et al., Civ. A. No. 2:16-cv-02562 (D. Nev.). In November 2016, Amarin filed a lawsuit against Teva Pharmaceuticals USA, Inc. and Teva Pharmaceuticals Industries Limited (collectively, “Teva”) in the U.S. District Court for the District of Nevada. The case against Teva is captioned Amarin Pharma, Inc. et al. v. Teva Pharmaceuticals USA, Inc. et al., Civ. A. No. 2:16-cv-02658. In all three lawsuits, Amarin is seeking, among other remedies, an order enjoining each defendant from marketing generic versions of Vascepa
64
before the last to expire of the asserted patents in 2030. The three lawsuits have been consolidated for pretrial proceedings. As a result of the statutory stay associated with the filing of these lawsuits under the Hatch-Waxman Act, the FDA cannot grant final approval to West-Ward, DRL, or Teva’s respective ANDA before January 2020, unless there is an earlier court decision holding that the subject patents are not infringed and/or are invalid.
The fourth ANDA applicant referenced above is Apotex Inc. (“Apotex”), which sent Amarin a paragraph IV certification notice in September 2016. The notice reflected that Apotex made a paragraph IV notice as to some, but not all, of the patents listed in the Orange Book for Vascepa. Because Apotex did not make a paragraph IV certification as to all listed patents, Apotex cannot market a generic version of Vascepa before the last to expire of the patents for which Apotex did not make a paragraph IV certification, which is in 2030. At a later date, Apotex may elect to amend its ANDA in order to make a paragraph IV certification as to additional listed patents. If and when Apotex does make such an amendment, it would be required to send Amarin an additional paragraph IV certification notice, and Amarin would then have the ability to file a lawsuit against Apotex pursuant to the Hatch-Waxman Act.
In October 2016, we introduced to the market a 0.5-gram dose strength of Vascepa. In August 2017, as anticipated, we received a paragraph IV certification notice from Teva contending that certain of our patents are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of the 0.5-gram dose strength of Vascepa, as described in the Teva ANDA. This Teva ANDA was filed as an amendment to the 1-gram Teva ANDA and is related to patents already at issue in the 1-gram Vascepa patent litigation. This certification followed the related listing in the Orange Book of patents associated with the 0.5-gram product in June 2017. This June 2017 listing was within the five-year, post NDA-approval period during which the Hatch-Waxman Amendments require a paragraph IV certification of patent invalidity or non-infringement under the Hatch-Waxman, five-year, NCE regulatory scheme. Accordingly, in October 2017, we filed a patent infringement lawsuit against Teva in the U.S. District Court for the District of Nevada. The case is captioned Amarin Pharma, Inc. et al. v. Teva Pharmaceuticals USA, Inc. et al., Civ. A. No. 2:17-cv-2641 (D. Nev.). In this lawsuit, we sought, among other remedies, an order enjoining Teva from marketing generic versions of the 0.5-gram dose strength of Vascepa before the last to expire of the asserted patents in 2030.
On May 24, 2018, we entered into a settlement agreement with Teva that resolves our ANDA patent litigation as it relates to Teva’s as amended ANDA for both the 1-gram and 0.5-gram dose strengths of Vascepa. As part of this settlement agreement, Teva may first begin selling its generic version of Vascepa in the United States on August 9, 2029, or earlier under certain customary circumstances, including commercial launch by another generic manufacturer under certain circumstances, in which event Teva would pay us certain royalties on its generic Vascepa products. The ANDA patent litigation continues in the United States District Court for the District of Nevada with parties West-Ward and DRL.
In July 2018, as anticipated, we received a paragraph IV certification notice from DRL contending that certain of our patents are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of the 0.5-gram dose strength of Vascepa, as described in the DRL ANDA. This DRL ANDA was filed as an amendment to the 1-gram DRL ANADA and is related to patents already at issue in the 1-gram Vascepa patent litigation. This certification followed the related listing in the Orange Book of patents associated with the 0.5-gram product in June 2017. This June 2017 listing was within the five-year, post NDA-approval period during which the Hatch-Waxman Amendments require a paragraph IV certification of patent invalidity or non-infringement lawsuit against DRL in the U.S. District Court for the District of Nevada. The case is captioned Amarin Pharma, Inc. et al. v. Dr. Reddy’s Laboratories, Inc. et al., Civ. A. No. 2:18-cv-01596 (D. Nev.). In this lawsuit, we are seeking, among other remedies, an order enjoining DRL from marketing generic versions of the 0.5-gram dose strength of Vascepa before the last to expire of the asserted patents in 2030. In light of the overlap between the cases, DRL and Amarin have stipulated that the final judgment on the merits of the parties’ contentions in the consolidated 1-gram action shall also be binding in the 0.5-gram case.
We intend to vigorously enforce our intellectual property rights relating to Vascepa, but we cannot predict the outcome of these lawsuits or any subsequently filed lawsuits.
In addition to the above, in the ordinary course of business, we are from time to time involved in lawsuits, claims, investigations, proceedings, and threats of litigation relating to intellectual property, commercial arrangements and other matters.
Not applicable.
65
|
Item 5. |
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
The following table sets forth the high and low prices for our ADSs in each of the quarters over the past two fiscal years, as quoted on The NASDAQ Global Market under the symbol “AMRN.”
|
|
|
Common Stock Price |
|
|||||||||||||
|
|
Fiscal 2018 |
|
|
Fiscal 2017 |
|
|||||||||||
|
|
|
High |
|
|
Low |
|
|
High |
|
|
Low |
|
||||
|
First Quarter |
|
$ |
4.60 |
|
|
$ |
2.91 |
|
|
$ |
3.58 |
|
|
$ |
2.81 |
|
|
Second Quarter |
|
$ |
3.52 |
|
|
$ |
2.66 |
|
|
$ |
4.10 |
|
|
$ |
2.85 |
|
|
Third Quarter |
|
$ |
16.34 |
|
|
$ |
2.35 |
|
|
$ |
4.47 |
|
|
$ |
2.97 |
|
|
Fourth Quarter |
|
$ |
23.34 |
|
|
$ |
11.78 |
|
|
$ |
4.24 |
|
|
$ |
3.04 |
|
Shareholders
As of January 31, 2019, there were approximately 360 holders of record of our ordinary shares. Because many ordinary shares are held by broker nominees, we are unable to estimate the total number of shareholders represented by these record holders. Our depositary, Citibank, N.A., constitutes a single record holder of our ordinary shares.
Dividends
We have never paid dividends on common shares and do not anticipate paying any cash dividends on the common shares in the foreseeable future. Under English law, any payment of dividends would be subject to relevant legislation and our Articles of Association, which requires that all dividends must be approved by our Board of Directors and, in some cases, our stockholders, and may only be paid from our distributable profits available for the purpose, determined on an unconsolidated basis.
Under our Purchase and Sale Agreement with CPPIB Credit Europe S.à r.l., or CPPIB, as successor in interest to BioPharma Secured Debt Fund II Holdings Cayman LP, we are restricted from paying a dividend on our common shares, unless we have cash and cash equivalents in excess of a specified amount after such payment.
Performance Graph—5 Year
The following performance graph and related information shall not be deemed “soliciting material” or to be “filed” with the Securities and Exchange Commission, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing.
The following graph compares the cumulative 5-year return provided to stockholders of Amarin’s ADSs relative to the cumulative total returns of the NASDAQ Composite Index and the NASDAQ Biotechnology Index. We believe these indices are the most appropriate indices against which the total shareholder return of Amarin should be measured. The NASDAQ Biotechnology Index has been selected because it is an index of U.S. quoted biotechnology and pharmaceutical companies. An investment of $100 (with reinvestment of all dividends) is assumed to have been made in our ADSs and in each of the indices on January 1, 2014 and its relative performance is tracked through December 31, 2018.
66
Included in this 5-year time period is the substantial positive impact on the price of Amarin’s ADSs following presentation and publication of positive REDUCE-IT results in 2018.
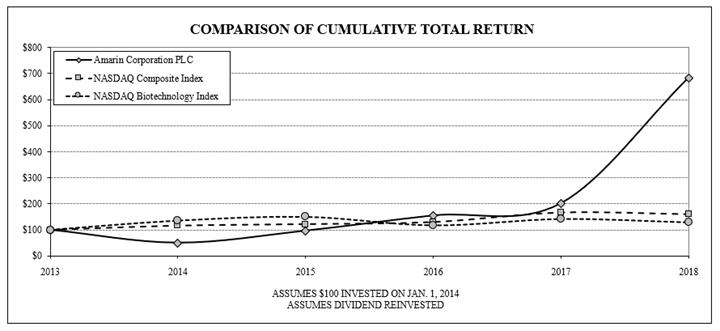
|
Company/Market/Peer Company |
|
12/31/2014 |
|
|
12/31/2015 |
|
|
12/31/2016 |
|
|
12/31/2017 |
|
|
12/31/2018 |
|
|||||
|
Amarin Corporation PLC |
|
$ |
50.25 |
|
|
$ |
96.48 |
|
|
$ |
154.77 |
|
|
$ |
201.51 |
|
|
$ |
683.92 |
|
|
NASDAQ Composite Index |
|
$ |
116.34 |
|
|
$ |
122.18 |
|
|
$ |
130.28 |
|
|
$ |
167.08 |
|
|
$ |
160.59 |
|
|
NASDAQ Biotechnology Index |
|
$ |
135.80 |
|
|
$ |
150.53 |
|
|
$ |
117.41 |
|
|
$ |
142.13 |
|
|
$ |
128.88 |
|
Performance Graph—3 Year
The following performance graph and related information shall not be deemed “soliciting material” or to be “filed” with the Securities and Exchange Commission, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing.
67
The following graph compares the cumulative 3-year return provided to stockholders of Amarin’s ADSs relative to the cumulative total returns of the NASDAQ Composite Index and the NASDAQ Biotechnology Index. We believe these indices are the most appropriate indices against which the total shareholder return of Amarin should be measured. The NASDAQ Biotechnology Index has been selected because it is an index of U.S. quoted biotechnology and pharmaceutical companies. An investment of $100 (with reinvestment of all dividends) is assumed to have been made in our ADSs and in each of the indices on January 1, 2016 and its relative performance is tracked through December 31, 2018.
Included in this 3-year time period is the substantial positive impact on the price of Amarin’s ADSs in 2018 following presentation and publication of positive REDUCE-IT results. During this entire 3-year time period, cumulative total return for Amarin’s ADSs exceeded both the NASDAQ Composite Index and NASDAQ Biotechnology Index.

|
Company/Market/Peer Company |
|
12/31/2016 |
|
|
12/31/2017 |
|
|
12/31/2018 |
|
|||
|
Amarin Corporation PLC |
|
$ |
162.96 |
|
|
$ |
212.17 |
|
|
$ |
720.11 |
|
|
NASDAQ Composite Index |
|
$ |
107.50 |
|
|
$ |
137.86 |
|
|
$ |
132.51 |
|
|
NASDAQ Biotechnology Index |
|
$ |
78.32 |
|
|
$ |
94.81 |
|
|
$ |
85.97 |
|
Information about Our Equity Compensation Plans
Information regarding our equity compensation plans is incorporated by reference in Item 12 of Part III of this annual report on Form 10-K.
Unregistered Sales of Equity Securities and Use of Proceeds
Issuer Purchases of Equity Securities
Shares purchased in the fourth quarter of 2018 are as follows:
|
Period |
|
Total Number of Shares Purchased (1) |
|
|
Average Price Paid per Share |
|
||
|
October 1 – 31, 2018 |
|
|
— |
|
|
$ |
— |
|
|
November 1 – 30, 2018 |
|
|
— |
|
|
|
— |
|
|
December 1 – 31, 2018 |
|
|
41,252 |
|
|
|
13.24 |
|
|
Total |
|
|
41,252 |
|
|
$ |
13.24 |
|
|
(1) |
Represents shares withheld to satisfy tax withholding amounts due from employees related to the receipt of stock which resulted from the exercise or vesting of equity awards. |
68
Capital Gains
If you are not resident in the United Kingdom, or UK, for UK tax purposes, you will not be liable for UK tax on capital gains realized or accrued on the sale or other disposition of common shares or ADSs unless the common shares or ADSs are held in connection with your trade carried on in the UK through a branch or agency and the common shares or ADSs are or have been used, held or acquired for the purposes of such trade or such branch or agency.
An individual holder of common shares or ADSs who ceases to be resident in the UK for UK tax purposes for a period of less than 5 years and who disposes of common shares or ADSs during that period may also be liable on returning to the UK for UK capital gains tax despite the fact that the individual may not be resident in the UK at the time of the disposal.
Inheritance Tax
If you are an individual domiciled in the United States and are not a national of the UK for the purposes of the Inheritance and Gift Tax Treaty 1978 between the United States and the UK, any common shares or ADSs beneficially owned by you will not generally be subject to UK inheritance tax on your death or on a gift made by you during your lifetime, provided that any applicable United States federal gift or estate tax liability is paid, except where the common share or ADS is part of the business property of your UK permanent establishment.
Where the common shares or ADSs have been placed in trust by a settlor who, at the time of the settlement, was domiciled in the United States and not a national of the UK, the common shares or ADSs will not generally be subject to UK inheritance tax.
Stamp Duty and Stamp Duty Reserve Tax
Transfer of ADSs
No UK stamp duty will be payable on an instrument transferring an ADS or on a written agreement to transfer an ADS provided that the instrument of transfer or the agreement to transfer is executed and remains at all times outside the UK. Where these conditions are not met, the transfer of, or agreement to transfer, an ADS could, depending on the circumstances, attract a charge to ad valorem stamp duty at the rate of 0.5% of the value of the consideration.
No stamp duty reserve tax will be payable in respect of an agreement to transfer an ADS, whether made in or outside the UK.
Issuance and Transfer of Common Shares
The issuance of common shares by Amarin will not give rise to a charge to UK stamp duty or stamp duty reserve tax under current UK and European Union law; it is not currently known whether this position will continue for UK stamp duty reserve tax in relation to the issuance of common shares in return for an issuance of ADSs after the United Kingdom leaves the European Union. In the event of a change in this position resulting in the issuance of common shares by Amarin giving rise to a charge to UK stamp duty or stamp duty reserve tax, Amarin would be responsible for any such UK stamp duty reserve tax payable on the issuance of common shares in return for the issuance of ADSs.
Transfers of common shares, as opposed to ADSs, will attract ad valorem stamp duty at the rate of 0.5% of the amount or value of the consideration. A charge to stamp duty reserve tax, at the rate of 0.5% of the amount or value of the consideration, will arise on an agreement to transfer common shares. The stamp duty reserve tax is payable on the seventh day of the month following the month in which the charge arises. Where an instrument of transfer is executed and duly stamped before the expiry of a period of six years beginning with the date of that agreement, any stamp duty reserve tax that has not been paid ceases to be payable.
Taxation of Dividends
Under UK law, there is no withholding tax on dividends paid on the common shares or ADSs.
69
The selected financial data set forth below as of and for the years ended December 31, 2018, 2017, 2016, 2015, and 2014 have been derived from the audited consolidated financial statements of Amarin. This data should be read in conjunction with our audited consolidated financial statements and related notes which are included elsewhere in this Annual Report on Form 10-K, and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in Item 7 below. Historical results are not necessarily indicative of operating results to be expected in the future.
|
|
|
|
Years Ended December 31, |
|
|||||||||||||||||
|
|
|
|
2018 |
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||||
|
|
|
|
(In thousands, except per share amounts) |
|
|||||||||||||||||
|
Consolidated Statements of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product revenue, net |
|
$ |
228,371 |
|
|
$ |
179,825 |
|
|
$ |
128,966 |
|
|
$ |
80,987 |
|
|
$ |
54,202 |
|
|
|
Licensing revenue |
|
|
843 |
|
|
|
1,279 |
|
|
|
1,118 |
|
|
|
769 |
|
|
|
— |
|
|
|
Total revenue, net |
|
|
229,214 |
|
|
|
181,104 |
|
|
|
130,084 |
|
|
|
81,756 |
|
|
|
54,202 |
|
|
|
Less: Cost of goods sold |
|
|
54,543 |
|
|
|
44,952 |
|
|
|
34,363 |
|
|
|
27,875 |
|
|
|
20,485 |
|
|
|
Gross margin |
|
|
174,671 |
|
|
|
136,152 |
|
|
|
95,721 |
|
|
|
53,881 |
|
|
|
33,717 |
|
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Selling, general and administrative (1) |
|
|
226,996 |
|
|
|
134,549 |
|
|
|
111,372 |
|
|
|
101,041 |
|
|
|
79,346 |
|
|
|
Research and development |
|
|
55,900 |
|
|
|
47,158 |
|
|
|
49,975 |
|
|
|
51,062 |
|
|
|
50,326 |
|
|
|
Total operating expenses |
|
|
282,896 |
|
|
|
181,707 |
|
|
|
161,347 |
|
|
|
152,103 |
|
|
|
129,672 |
|
|
|
Operating loss |
|
|
(108,225 |
) |
|
|
(45,555 |
) |
|
|
(65,626 |
) |
|
|
(98,222 |
) |
|
|
(95,955 |
) |
|
|
Gain (loss) on change in fair value of derivative liabilities (2) |
|
|
— |
|
|
|
— |
|
|
|
8,170 |
|
|
|
(1,106 |
) |
|
|
13,472 |
|
|
|
Gain on extinguishment of debt |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,314 |
|
|
|
38,034 |
|
|
|
Interest expense |
|
|
(8,872 |
) |
|
|
(9,766 |
) |
|
|
(18,677 |
) |
|
|
(20,180 |
) |
|
|
(18,575 |
) |
|
|
Interest income |
|
|
1,074 |
|
|
|
429 |
|
|
|
234 |
|
|
|
132 |
|
|
|
96 |
|
|
|
Other (expense) income, net |
|
|
(326 |
) |
|
|
74 |
|
|
|
(482 |
) |
|
|
(228 |
) |
|
|
3,727 |
|
|
|
Loss from operations before taxes |
|
|
(116,349 |
) |
|
|
(54,818 |
) |
|
|
(76,381 |
) |
|
|
(118,290 |
) |
|
|
(59,201 |
) |
|
|
(Provision for) benefit from income taxes (5) |
|
|
(96 |
) |
|
|
(13,047 |
) |
|
|
(9,969 |
) |
|
|
3,086 |
|
|
|
2,837 |
|
|
|
Net loss |
|
|
(116,445 |
) |
|
|
(67,865 |
) |
|
|
(86,350 |
) |
|
|
(115,204 |
) |
|
|
(56,364 |
) |
|
|
Preferred stock purchase option |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(868 |
) |
|
|
— |
|
|
|
Preferred stock beneficial conversion features |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(32,987 |
) |
|
|
— |
|
|
|
Net loss applicable to common shareholders |
|
|
(116,445 |
) |
|
|
(67,865 |
) |
|
|
(86,350 |
) |
|
|
(149,059 |
) |
|
|
(56,364 |
) |
|
|
Loss per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
(0.39 |
) |
|
$ |
(0.25 |
) |
|
$ |
(0.41 |
) |
|
$ |
(0.83 |
) |
|
$ |
(0.32 |
) |
|
|
Diluted |
|
$ |
(0.39 |
) |
|
$ |
(0.25 |
) |
|
$ |
(0.41 |
) |
|
$ |
(0.83 |
) |
|
$ |
(0.36 |
) |
|
|
Weighted average shares: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
|
297,237 |
|
|
|
270,652 |
|
|
|
211,874 |
|
|
|
180,654 |
|
|
|
173,719 |
|
|
|
Diluted |
|
|
297,237 |
|
|
|
270,652 |
|
|
|
211,874 |
|
|
|
180,654 |
|
|
|
173,824 |
|
|
|
|
|
|
As of December 31, |
|
|||||||||||||||||
|
|
|
|
2018 |
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||||
|
|
|
|
(In thousands) |
|
|||||||||||||||||
|
Consolidated Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
Cash and cash equivalents |
|
$ |
249,227 |
|
|
$ |
73,637 |
|
|
$ |
98,251 |
|
|
$ |
106,961 |
|
|
$ |
119,539 |
|
|
|
Total assets (3) (4) |
|
|
385,714 |
|
|
|
161,598 |
|
|
|
166,999 |
|
|
|
173,230 |
|
|
|
168,886 |
|
|
|
Long-term liabilities (3) |
|
|
76,121 |
|
|
|
118,168 |
|
|
|
99,808 |
|
|
|
250,059 |
|
|
|
217,028 |
|
|
|
Stockholders’ equity (deficit) (4) |
|
|
152,330 |
|
|
|
(65,100 |
) |
|
|
(9,058 |
) |
|
|
(127,552 |
) |
|
|
(88,448 |
) |
|
|
(1) |
Includes non-cash warrant-related compensation income in 2014 and 2015, reflecting the change in the fair value of the warrant derivative liability associated with warrants issued in October 2009 to former officers of Amarin. |
|
(2) |
Includes non-cash charges resulting from changes in the fair value of derivative liabilities. |
70
|
(4) |
Reflects recognition of deferred tax assets of approximately $1.6 million relating to excess tax benefits on stock-based compensation outstanding as of December 31, 2015 and corresponding cumulative-effect adjustment to accumulated deficit as of December 31, 2015, due to the modified retrospective application of ASU No. 2016-09, Compensation—Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting, adopted in 2016. |
This Annual Report on Form 10-K contains forward-looking statements concerning future events and performance of the Company. When used in this report, the words “may,” “would,” “should,” “could,” “expects,” “aims,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “projects,” “potential,” or “continue” or the negative of these terms or other comparable terminology are included to identify forward-looking statements. These statements include but are not limited to statements regarding the commercial success of Vascepa and factors that can affect such success; interpretation of court decisions; expectation on determinations and policy positions of the United States Food and Drug Administration, or FDA; the expected timing of enrollment, interim results and final results of our REDUCE-IT study; the safety and efficacy of our product and product candidates; expectation regarding the potential for Vascepa to be partnered, developed and commercialized outside of the United States; expectation on the scope and strength of our intellectual property protection and the likelihood of securing additional patent protection; estimates of the potential markets for our product candidates; estimates of the capacity of manufacturing and other facilities to support our products; our operating and growth strategies; our industry; our projected cash needs, liquidity and capital resources; and our expected future revenues, operations and expenditures. These forward-looking statements are based on our current expectations and assumptions and many factors could cause our actual results to differ materially from those indicated in these forward-looking statements. You should review carefully the factors identified in this report in Item 1A, “Risk Factors”. We disclaim any intent to update or announce revisions to any forward-looking statements to reflect actual events or developments, except as required by law. Except as otherwise indicated herein, all dates referred to in this report represent periods or dates fixed with reference to our fiscal year ended December 31, 2018.
We are a pharmaceutical company with expertise in omega-3 fatty acids and lipid science focused on the commercialization and development of therapeutics to improve cardiovascular health and reduce cardiovascular, or CV, risk.
71
Our lead product, Vascepa® (icosapent ethyl) capsules, is approved by the U.S. Food and Drug Administration, or FDA, for use as an adjunct to diet to reduce triglyceride, or TG, levels in adult patients with severe (TG ≥500 mg/dL) hypertriglyceridemia. In January 2013, we began selling and marketing Vascepa in the United States by prescription only based on the FDA-approved MARINE indication of patients with severe (TG ≥500 mg/dL) triglyceride levels. We sell 1-gram and 0.5-gram capsule sizes of Vascepa principally to a limited number of major wholesalers, as well as selected regional wholesalers and specialty pharmacy providers, or collectively, our Distributors or our customers, that in turn resell Vascepa to retail pharmacies for subsequent resale to patients and healthcare providers.
Since our inception, we have devoted substantial resources to our research and development efforts, most significantly our Vascepa cardiovascular outcomes trial, REDUCE-IT. We announced topline results from the REDUCE-IT study on September 24, 2018. On November 10, 2018, we publicly presented primary results of the REDUCE-IT study at the 2018 Scientific Sessions of the American Heart Association, or AHA, and the results were concurrently published in The New England Journal of Medicine. REDUCE-IT met its primary endpoint demonstrating a 25% relative risk reduction, or RRR, to a high degree of statistical significance (p<0.001), in first occurrence of major adverse cardiovascular events, or MACE, in the intent-to-treat patient population with use of Vascepa 4 grams/day as compared to placebo. REDUCE-IT also showed a 26% RRR in its key secondary composite endpoint of cardiovascular death, heart attacks and stroke (p<0.001). Based on the final positive results of REDUCE-IT, we plan to seek additional indicated uses for Vascepa in the United States and to continue to develop Vascepa commercially in major markets around the world. We anticipate submitting a supplemental new drug application (sNDA) to the FDA before the end of March 2019 based on the positive results of the REDUCE-IT study.
We promote Vascepa directly in the United States. Such promotion, prior to results of the REDUCE-IT study, was based on demonstrated changes in biomarkers based on our MARINE and ANCHOR studies. Most healthcare professionals express that they prefer outcomes data to biomarker data. Because prior to results of the REDUCE-IT study we did not have outcomes data regarding the clinical effect of Vascepa and because a substantial portion of our resources were being spent on the REDUCE-IT study, prior to REDUCE-IT results our commercialization of Vascepa was somewhat limited. Subsequent to learning the positive cardiovascular outcomes results of the REDUCE-IT study, we have begun increasing our promotion of Vascepa.
In addition to promotion of Vascepa in the United States, we have entered into strategic partnerships and license arrangements in Asia, the Middle East, North Africa and Canada to further promote, develop and commercialize Vascepa. We continue to assess other collaboration opportunities to maximize the value of the Vascepa franchise globally.
Commercialization—United States
We commenced the commercial launch of 1-gram size Vascepa capsules in the United States in January 2013. We commenced sales and shipments of Vascepa at that time to our network of U.S.-based wholesalers. Prior to REDUCE-IT results topline announcement in September 2018, our direct sales force consisted of approximately 170 sales professionals, including sales representatives and their managers. We have recently increased the size of our sales force to approximately 440 sales professionals, including approximately 400 sales representatives, pursuant to positive REDUCE-IT results and are expanding our promotion of Vascepa. Commencing in May 2014, in addition to Vascepa promotion by our sales representatives, Kowa Pharmaceuticals America, Inc. began co-promoting Vascepa in conjunction with its promotion of its primary product, a branded statin for patients with high cholesterol. Amarin and Kowa Pharmaceuticals America, Inc. intentionally designed the co-promotion to naturally end as of December 31, 2018 and mutually agreed not to renew the agreement. We also employ various medical affairs and marketing personnel to support our commercialization of Vascepa. We expanded certain medical education and market awareness initiatives following the reporting of positive REDUCE-IT results in 2018. We intend to further expand promotion of Vascepa following label expansion of Vascepa, subject to FDA approval of such expanded label.
In October 2016, in addition to the original 1-gram capsule size for Vascepa, we introduced a smaller 0.5-gram capsule size, the first and only 0.5-gram prescription omega-3 alternative available on the market, for the subset of patients who prefer a smaller capsule. The FDA-approved dosing for Vascepa continues to be 4 grams per day, and, as expected, the majority of new and existing patients taking Vascepa continue to be prescribed the 1-gram size Vascepa capsule.
72
From May 2014 until December 2018, under our co-promotion agreement with Kowa Pharmaceuticals America, Inc., both parties agreed to use commercially reasonable efforts to promote, detail and optimize sales of Vascepa in the United States and agreed to specific performance requirements detailed in the related agreement. The performance requirements included a negotiated minimum number of sales details to be delivered by each party in the first and second position, the use of a negotiated number of minimum sales representatives from each party, and the achievement of minimum levels of Vascepa revenue in 2015 and beyond. First position referred to when a sales representative’s primary purpose in detailing is related to Vascepa, while second position referred to when a sales representative’s primary purpose in detailing is to promote another product, but they also devoted time in the same sales call to promote Vascepa. Kowa Pharmaceuticals America, Inc. also agreed to bear the costs incurred for its sales force associated with the commercialization of Vascepa and to pay for certain incremental costs associated with the use of its sales force, such as sample costs and costs for promotional and marketing materials. We recognized all revenue from sales of Vascepa. In exchange for Kowa Pharmaceuticals America, Inc.’s co-promotional services, Kowa Pharmaceuticals America, Inc. was entitled to a quarterly co-promotion fee based on a percentage of aggregate Vascepa gross margin that varied during the term. The percentage of aggregate Vascepa gross margin earned by Kowa Pharmaceuticals America, Inc. was, as amended, approximately eighteen percent (18%) in 2017, partially offset by certain other refinements. During 2018, which was the last year of the agreement, as amended, we incurred expense for both the annual co-promotion fee, which in 2018 was calculated as eighteen-and-a-half percent (18.5%) of Vascepa gross margin, plus accrual for co-promotion tail payments which were calculated as a percentage of the 2018 co-promotion fee. Kowa Pharmaceuticals America, Inc. is eligible to receive $17.8 million in co-promotion tail payments, the present value of which of $16.6 million was fully accrued as of December 31, 2018. The accrued tail payments will be paid over three years with declining amounts each year beginning with $7.3 million to be paid in 2019.
Based on monthly compilations of data provided by a third party, Symphony Health, the estimated number of normalized total Vascepa prescriptions for the three months ended December 31, 2018 was approximately 539,000 compared to 458,000, 430,000, 391,000, and 404,000 in the three months ended September 30, 2018, June 30, 2018, March 31, 2018, and December 31, 2017, respectively. According to data from another third party, IQVIA, the estimated number of normalized total Vascepa prescriptions for the three months ended December 31, 2018 was approximately 538,000 compared to 457,000, 430,000, 392,000, and 409,000 in the three months ended September 30, 2018, June 30, 2018, March 31, 2018, and December 31, 2017, respectively. Normalized total prescriptions represent the estimated total number of Vascepa prescriptions dispensed to patients, calculated on a normalized basis (i.e., one month’s supply, or total capsules dispensed multiplied by the number of grams per capsule divided by 120 grams). Inventory levels at wholesalers tend to fluctuate based on seasonal factors, prescription trends and other factors.
The data reported above is based on information made available to us from third-party resources and may be subject to adjustment and may overstate or understate actual prescriptions. Timing of shipments to wholesalers, as used for revenue recognition purposes, and timing of prescriptions as estimated by these third parties may differ from period to period. Although we believe these data are prepared on a period-to-period basis in a manner that is generally consistent and that such results can be generally indicative of current prescription trends, these data are based on estimates and should not be relied upon as definitive. While we expect to be able to grow Vascepa revenues over time, no guidance should be inferred from the operating metrics described above. We also anticipate that such sales growth will be inconsistent from period to period. We believe that investors should view the above-referenced operating metrics with caution, as data for this limited period may not be representative of a trend consistent with the results presented or otherwise predictive of future results. Seasonal fluctuations in pharmaceutical sales, for example, may affect future prescription trends of Vascepa, as could changes in prescriber sentiment, quarterly changes in Distributor purchases, and other factors. We believe investors should consider our results over several quarters, or longer, before making an assessment about potential future performance.
Commercialization—Outside the United States
In February 2015, we announced an exclusive agreement with Eddingpharm to develop and commercialize Vascepa capsules in what we refer to as the China Territory, consisting of the territories of Mainland China, Hong Kong, Macau and Taiwan, for uses that are currently commercialized and under development by us in the United States based on the MARINE, ANCHOR and REDUCE-IT clinical trials of Vascepa. Under the agreement, Eddingpharm is responsible for development and commercialization activities in the China Territory and associated expenses. We will provide development assistance and be responsible for supplying the product. Terms of the agreement include up-front and milestone payments to us of up to $169.0 million, including a non-refundable $15.0 million up-front payment received at closing and a non-refundable milestone payment of $1.0 million received upon successful submission of a clinical trial application, or CTA, with respect to the MARINE indication for Vascepa to the Chinese regulatory authority in March 2016. In March 2017, the CTA was approved by the Chinese regulatory authority and, in December 2017, Eddingpharm commenced a pivotal clinical trial aimed to support the regulatory approval of the first indication of Vascepa in a patient population with severe hypertriglyceridemia in Mainland China. We are also entitled to receive future regulatory and sales-based milestone payments of up to an additional $153.0 million. The regulatory milestone events relate to the submission and approval of certain applications to the applicable regulatory authority, such as a clinical trial application, clinical trial exemption, or import drug license application. The amounts to be received upon achievement of the regulatory milestone events relate to the submission and approval for three indications, and range from $2.0 million to $15.0 million for a total of $33.0 million. The sales-based milestone events occur when annual aggregate net sales of Vascepa in the territory equals or exceeds certain specified thresholds, and range from $5.0 million to
73
$50.0 million for a total of $120.0 million. Eddingpharm will also pay us tiered double-digit percentage royalties on net sales of Vascepa in the China Territory escalating to the high teens. We will supply finished product to Eddingpharm under negotiated terms.
In March 2016, we entered into an agreement with Biologix FZCo, or Biologix, to register and commercialize Vascepa in several Middle Eastern and North African countries. Under the terms of the distribution agreement, we granted to Biologix a non-exclusive license to use our trademarks in connection with the importation, distribution, promotion, marketing and sale of Vascepa in the Middle East and North Africa territory. Upon closing of the agreement, we received a non-refundable up-front payment, which will be recognized as revenue over 10 years commencing upon first marketing approval of Vascepa in the territory. We receive all payments based on total product sales and pay Biologix a service fee in exchange for its services, whereby the service fee represents a percentage of gross selling price which is subject to a minimum floor price. In March 2018 and July 2018, we received approval for Vascepa as a prescription medication for use in Lebanon and United Arab Emirates, respectively, as an adjunct to diet to reduce triglyceride levels in adult patients with severe hypertriglyceridemia.
In September 2017, we entered into an agreement with HLS to register, commercialize and distribute Vascepa in Canada. Under the agreement, HLS will be responsible for regulatory and commercialization activities and associated costs. We will be responsible for providing assistance towards local filings, supplying finished product under negotiated supply terms, maintaining intellectual property, and continuing the development and funding of REDUCE-IT related activities. Terms of the agreement include up-front and milestone payments to us of up to $65.0 million. These payments include a non-refundable $5.0 million up-front payment received in two equal installments, the first of which was received at closing with the second received upon the six-month anniversary of the closing, as well as a non-refundable milestone payment of $2.5 million received upon achievement of the REDUCE-IT trial primary endpoint. In addition to the non-refundable, up-front payment, we are entitled to receive certain regulatory and sales-based milestone payments of up to an additional $57.5 million, the timing and achievability of which cannot be determined at least until discussions with Canadian regulatory authorities have commenced, as well as tiered double-digit royalties on net sales of Vascepa in Canada.
We continue to assess other partnership opportunities for licensing Vascepa to partners outside of the United States.
Since its inception in 2011, conduct of the REDUCE-IT cardiovascular outcomes study of Vascepa has been the centerpiece of our research and development. Most of our other research and development during this period also pertained to Vascepa, including study of the mechanism of action of the single active ingredient in Vascepa, icosapent ethyl. The REDUCE-IT study, since its inception in 2011, was conducted based on a special protocol assessment, or SPA, agreement with the FDA. Based on the final positive results of REDUCE-IT, we plan to seek additional indicated uses for Vascepa in the United States and to pursue approval for Vascepa around the world. We also anticipate continuing to publish additional details of the REDUCE-IT study to address scientific interest beyond the primary results of this study derived from the over 35,000 patient years of study experience which were accumulated in the REDUCE-IT study. The REDUCE-IT study topline results were made public in September 2018, and the primary results of the REDUCE-IT study were presented at the 2018 Scientific Sessions of the American Heart Association (AHA) on November 10, 2018 with such results concurrently published in The New England Journal of Medicine. Potential additional research and development opportunities beyond REDUCE-IT will be prioritized after giving priority to securing regulatory approval for Vascepa based on the REDUCE-IT results in the United States and in various geographies internationally, including pursuit of approval for Vascepa in Europe and in countries where we have commercialization partners for Vascepa. The indication we intend to seek based on the final positive results of REDUCE-IT pertains to use of Vascepa to reduce cardiovascular events in at-risk patients. While the current FDA approved indication for Vascepa is biomarker based (i.e., lowering triglyceride levels), the indication we will seek based on REDUCE-IT results will be outcomes based (i.e., lowering cardiovascular events).
It is believed that the effects of EPA are not due to a single mode of action, such as triglyceride lowering, but rather to multiple mechanisms working together. Studies in the scientific literature explore potentially beneficial effects of EPA on multiple atherosclerosis processes, including endothelial function, oxidative stress, foam cell formation, inflammation/cytokines, plaque formation/progression, platelet aggregation, thrombus formation, and plaque rupture. With respect to triglyceride levels, our scientific rationale for the REDUCE-IT study was supported by (i) epidemiological data that suggests elevated triglyceride levels correlate with increased cardiovascular disease risk, (ii) genetic data that suggests triglyceride and/or triglyceride-rich lipoproteins (as well as low-density lipoprotein cholesterol (LDL cholesterol), known as bad cholesterol) are independently in the causal pathway for cardiovascular disease and (iii) clinical data that suggest substantial triglyceride reduction in patients with elevated baseline triglyceride levels correlates with reduced cardiovascular risk. The REDUCE-IT study was designed to determine the clinical benefit, if any, of stable EPA therapy in statin-treated patients with elevated triglyceride levels.
In June 2018, we entered into a multi-faceted collaboration with Mochida related to the development and commercialization of drug products and indications based on the active pharmaceutical ingredient in Vascepa, the omega-3 acid, EPA. Among other terms in the agreement, we obtained an exclusive license to certain Mochida intellectual property to advance our interests in the United States and certain other territories and the parties will collaborate to research and develop new products and indications based on EPA for our commercialization in the United States and certain other territories. The potential new product and indication opportunities
74
contemplated under this agreement are currently in early stages of development. Upon closing of the collaboration agreement, we made a non-refundable, non-creditable upfront payment of approximately $2.7 million. In addition, the agreement provides for milestone payments from us upon the achievement of certain product development milestones and royalties on net sales of future products arising from the collaboration, if any. Expenditures related to research and development activities for product candidates under the collaboration agreement were immaterial in 2018 and are expected to be less than $5.0 million in 2019.
Commercial and Clinical Supply
We manage the manufacturing and supply of Vascepa internally and have done so since we began clinical development of Vascepa prior to the drug’s marketing approval by FDA in 2012. We rely on contract manufacturers in each step of our commercial and clinical product supply chain. These steps include active pharmaceutical ingredient, or API, manufacturing, encapsulation of the API, product packaging and supply-related logistics. Our approach to product supply procurement is designed to mitigate risk of supply interruption and maintain an environment of cost competition through diversification of contract manufacturers at each stage of the supply chain and lack of reliance on any single supplier. We have multiple FDA-approved international API suppliers, encapsulators and packagers to support Vascepa commercial franchise. The amount of supply we seek to purchase in future periods will depend on the level of growth of Vascepa revenues and minimum purchase commitments with certain suppliers. While our current supply chain is scalable, we continue efforts to expand, diversify and further enhance it.
Financial Position
We believe that our cash and cash equivalents of $249.2 million as of December 31, 2018, will be sufficient to fund our projected operations for at least twelve months and through the likely Prescription Drug User Fee Act (PDUFA) date for approval of a sNDA by the FDA based on REDUCE-IT study results. Depending on the level of cash generated from operations, and depending in part on the timing and results of the FDA review of the sNDA and rate of prescription growth for Vascepa, additional capital may be required to support planned expansion of Vascepa promotion and potential Vascepa promotion beyond which we are currently executing. If additional capital is required and we are unable to obtain additional capital, we may be forced to delay, limit or eliminate certain promotional activities. We anticipate that quarterly net cash outflows in future periods will be variable.
Product Revenue, net. All of our product revenue is derived from product sales of 1-gram and 0.5-gram size capsules of Vascepa, net of allowances, discounts, incentives, rebates, chargebacks and returns. We sell product to a limited number of major wholesalers, as well as selected regional wholesalers and specialty pharmacy providers, or collectively, our Distributors or our customers, who resell the product to retail pharmacies for purposes of their reselling the product to fill patient prescriptions. We commenced our commercial launch of 1-gram size Vascepa capsules in the United States in January 2013, and introduced a smaller 0.5-gram capsule size in October 2016. Revenues from product sales are recognized when the Distributor obtains control of our product, which occurs at a point in time, typically upon delivery to the Distributor.
Licensing revenue. Licensing revenue currently consists of revenue attributable to receipt of up-front, non-refundable payments and milestone payments related to license and distribution agreements for Vascepa outside the United States. We recognize revenue from licensing arrangements as we fulfill the performance obligations under each of the agreements.
Cost of Goods Sold. Cost of goods sold includes the cost of API for Vascepa on which revenue was recognized during the period, as well as the associated costs for encapsulation, packaging, shipment, supply management, quality assurance, insurance, and other indirect manufacturing, logistics and product support costs. The cost of the API included in cost of goods sold reflects the average cost method of inventory valuation and relief. This average cost reflects the actual purchase price of Vascepa API.
Selling, General and Administrative Expense. Selling, general and administrative expense consists primarily of salaries and other related costs, including stock-based compensation expense, for personnel in our sales, marketing, executive, business development, finance and information technology functions, as well as co-promotion fees payable to Kowa Pharmaceuticals America, Inc. and, in 2018, the final year of the co-promotion agreement, accrual for the co-promotion tail payments. Other costs primarily include facility costs and professional fees for accounting, consulting and legal services.
Research and Development Expense. Research and development expense consists primarily of fees paid to professional service providers in conjunction with independent monitoring of our clinical trials and acquiring and evaluating data in conjunction with our clinical trials, fees paid to independent researchers, costs of qualifying contract manufacturers, services expenses incurred in developing and testing products and product candidates, salaries and related expenses for personnel, including stock-based compensation expense, costs of materials, depreciation, rent, utilities and other facilities costs. In addition, research and development expenses include the cost to support current development efforts, costs of product supply received from suppliers when such receipt by us is prior to regulatory approval of the supplier, as well as license fees related to our strategic collaboration with Mochida Pharmaceutical Co., Ltd. We expense research and development costs as incurred.
75
Gain on Change in Fair Value of Derivative Liabilities. Gain on change in fair value of derivative liabilities was comprised of: (i) the change in fair value of the derivative liability related to the change in control provision associated with the December 2012 royalty-bearing instrument financing arrangement, and (ii) the change in fair value of the derivative liabilities related to the change in control provisions associated with the previously outstanding May 2014 and November 2015 exchangeable senior notes.
Interest and Other (Expense) Income, Net. Interest expense consists of interest incurred under lease obligations, interest incurred under our December 2012 royalty-bearing instrument financing arrangement, and interest incurred under our previously outstanding 3.5% exchangeable notes. Interest expense under our royalty-bearing instrument financing arrangement is calculated based on an estimated repayment schedule. Interest expense under our exchangeable notes includes the amortization of the conversion option related to our exchangeable debt, the amortization of the related debt discounts and debt obligation coupon interest. Interest income consists of interest earned on our cash and cash equivalents. Other (expense) income, net, consists primarily of foreign exchange losses and gains.
Provision for Income Taxes. Provision for income taxes, deferred tax assets and liabilities, and reserves for unrecognized tax benefits reflect management’s best assessment of estimated future taxes to be paid. We are subject to income taxes in both the United States and foreign jurisdictions. The change in the effective tax rate was primarily driven by our evaluation of available evidence which resulted in the recording of non-cash provisions for income taxes against certain of the deferred tax assets for our U.S. subsidiary for the year ended December 31, 2017 and the continuation of that position for the year ended December 31, 2018.
Critical Accounting Policies and Significant Judgments and Estimates
Our discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements and notes, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue and expenses. On an ongoing basis, we evaluate our estimates and judgments, including those related to derivative financial liabilities. We base our estimates on historical experience and on various market-specific and other relevant assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. Estimates are assessed each period and updated to reflect current information. A summary of our significant accounting policies is contained in Note 2 to our consolidated financial statements included elsewhere in this Annual Report on Form 10-K. We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition—In accordance with GAAP, under Accounting Standards Codification (“ASC”) Topic 606, Revenue from Contracts with Customers, which we adopted on a modified retrospective basis effective January 1, 2018, we recognize revenue when our Distributors obtain control of promised goods or services, in an amount that reflects the consideration which we expect to receive in exchange for those goods or services. To determine revenue recognition for arrangements that we determine are within the scope of Topic 606, we perform the following five steps: (i) identify the contract(s) with a Distributor; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) we satisfy a performance obligation. We apply the five-step model to contracts only when it is probable that we will collect the consideration we are entitled to in exchange for the goods or services we transfer to the Distributor. At contract inception, once the contract is determined to be within the scope of Topic 606, we assess the goods or services promised within each contract, determine those that are performance obligations and assess whether each promised good or service is distinct. We then recognize as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied. For a complete discussion of our accounting for net product revenue and licensing revenues, see Note 2—Significant Accounting Policies.
We sell Vascepa principally to a limited number of Distributors that in turn resell Vascepa to retail pharmacies that subsequently resell it to patients and healthcare providers.
We began recognizing revenue from the sale of Vascepa following our commercial launch in the United States in January 2013. Prior to 2013, we recognized no revenue from Vascepa sales. In accordance with GAAP, we recognize revenue when the Distributor obtains control of our product, which occurs at a point in time, typically upon delivery to the Distributor. We recognized net product revenues of $228.4 million and $179.8 million based on sales to Distributors during the years ended December 31, 2018 and 2017, respectively.
We have written contracts with our Distributors, and transfer of control typically occurs upon delivery of our product to the Distributor. We evaluate the creditworthiness of each of our Distributors to determine whether revenues can be recognized upon delivery, subject to satisfaction of the other requirements, or whether recognition is required to be delayed until receipt of payment. We calculate gross product revenues based on the wholesale acquisition cost that we charge our Distributors for Vascepa. We estimate our net product revenues by deducting from our gross product revenues (a) trade allowances, such as invoice discounts for prompt payment and distributor fees, (b) estimated government and private payor rebates, chargebacks and discounts, such as Medicaid reimbursements,
76
(c) reserves for expected product returns and (d) estimated costs of incentives offered to certain indirect customers, including patients. The gross to net deductions are estimated based on available actual information, historical data, known trends, and levels of inventory in the distribution channel. We rely on resale data provided by our Distributors as well as prescription data provided by Symphony Health and IQVIA in estimating the level of inventory held in the distribution channel. A hypothetical 5% change in estimated aggregate bottles of channel inventory would result in a change of less than 1% in net product revenues reported during each of the three and twelve months ended December 31, 2018 and 2017.
When evaluating licensing arrangements, we perform the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) we satisfy each performance obligation. In determining performance obligations, we evaluate whether the license is distinct from the other performance obligations with the collaborative partner based on the consideration of the relevant facts and circumstances for each arrangement. Factors considered include the stage of development of the license delivered, research and development capabilities of the partner and the ability of partners to develop and commercialize Vascepa independent of us.
If the license to our intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, we recognize revenues from non-refundable, up-front fees allocated to the license when the license is transferred to the Distributor and the Distributor is able to use and benefit from the license. For licenses that are bundled with other promises, we utilize judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue from non-refundable, up-front fees. We evaluate the measure of progress each reporting period and, if necessary, adjust the measure of performance and related revenue recognition.
At the inception of each arrangement that includes development, regulatory and commercial milestone payments, we evaluate whether the milestones are considered probable of being reached and estimate the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within our control or the control of the licensee, such as regulatory approvals, are not considered probable of being achieved until those approvals are received. We evaluate factors such as the scientific, clinical, regulatory, commercial and other risks that must be overcome to achieve the respective milestone as well as the level of effort and investment required. The transaction price is then allocated to each performance obligation on a relative stand-alone selling price basis, for which we recognize revenue as or when the performance obligations under the contract are satisfied. At the end of each subsequent reporting period, we re-evaluate the probability of achievement of such development, regulatory and commercial milestones and any related constraint, and if necessary, adjust its estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect licensing revenues and earnings in the period of adjustment.
We receive payments from our customers based on billing schedules established in each contract. Up-front payments and fees are recorded as deferred revenue upon receipt or when due and may require deferral of revenue recognition to a future period until we perform our obligations under these arrangements. Amounts are recorded as accounts receivable when our right to consideration is unconditional. We do not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the customer and the transfer of the promised goods or services to the customer will be one year or less.
Inventory—We capitalize purchases of saleable inventory of Vascepa from suppliers that have been qualified by the FDA. The API purchased for Vascepa was sourced from three API suppliers in 2018, 2017, and 2016. If we add a new API supplier, all Vascepa API purchased from such supplier is included as a component of research and development expense until the new API supplier is approved. Upon sNDA approval of each additional supplier, we capitalize subsequent Vascepa API purchases from such supplier as inventory. We state inventories at the lower of cost or net realizable market value. Cost is determined based on actual cost using the average cost method. An allowance is established when management determines that certain inventories may not be saleable. If inventory cost exceeds expected net realizable value due to obsolescence, damage, quantities in excess of expected demand, changes in price levels or other causes, then we will reduce the carrying value of such inventory to net realizable value and recognize the difference as a component of cost of goods sold in the period in which it occurs. We expense inventory identified for use as marketing samples when they are packaged. The average cost reflects the actual purchase price of Vascepa API. Additionally, the determination of the classification of our inventory requires the use of estimates in order to determine the portion of inventories anticipated to be utilized within twelve months of the balance sheet date.
Income Taxes—Deferred tax assets and liabilities are recognized for the future tax consequences of differences between the carrying amounts and tax bases of assets and liabilities and operating loss carryforwards and other attributes using enacted rates
77
expected to be in effect when those differences reverse. Valuation allowances are provided against deferred tax assets that are not more likely than not to be realized.
We provide reserves for potential payments of tax to various tax authorities or do not recognize tax benefits related to uncertain tax positions and other issues. Tax benefits for uncertain tax positions are based on a determination of whether a tax benefit taken by us in our tax filings or positions is more likely than not to be realized, assuming that the matter in question will be decided based on its technical merits. Our policy is to record interest and penalties in the provision for income taxes.
We assess our ability to realize deferred tax assets at each reporting period. The realization of deferred tax assets depends on generating future taxable income during the periods in which the tax benefits are deductible or creditable. When making our assessment about the realization of our deferred tax assets as of December 31, 2018, we considered all available evidence, placing particular weight on evidence that could be objectively verified. The evidence considered included the (i) historical taxable profitability of our U.S. operations, (ii) historical and current year pre-tax book loss position, (iii) sources of future taxable income, giving weight to sources according to the extent to which they can be objectively verified, (iv) the provisions of the Tax Cuts and Jobs Act enacted in 2017 and their impact on our future taxable income, and (v) the risks to our business related to the commercialization and development of Vascepa. Based on our assessment, we concluded that all of our net deferred tax assets are not more likely than not to be realizable as of both December 31, 2018 and 2017. Changes in historical earnings performance, future earnings projections, and changes in tax laws and tax rates, among other factors, may cause us to adjust our valuation allowance on deferred tax assets in the future, which would impact our income tax expense in the period in which we determine that these factors have changed. We intend to maintain the valuation allowance until sufficient positive evidence exists to conclude that it is more likely than not that our deferred tax benefits will be realized. We will continue to monitor the need for valuation allowances in each jurisdiction and may adjust our positions in the future.
Excess tax benefits and deficiencies that arise upon vesting or exercise of share-based payments are recognized as an income tax benefit and expense, respectively, in the consolidated statement of operations.
Recent Accounting Pronouncements
For a discussion of recent accounting pronouncements, see Note 2—Significant Accounting Policies in the accompanying Notes to Consolidated Financial Statements in this Annual Report on Form 10-K.
Effects of Inflation
We believe the impact of inflation on operations has been minimal during the past three years.
Comparison of Fiscal Years Ended December 31, 2018 and December 31, 2017
Product Revenue, net. We recorded net product revenue of $228.4 million and $179.8 million during the years ended December 31, 2018 and 2017, respectively, an increase of $48.5 million, or 27%. This increase in revenue was driven primarily by an increase in estimated normalized total Vascepa prescriptions in the United States. Based on data provided by Symphony Health and IQVIA, estimated normalized total Vascepa prescriptions in the United States increased in 2018 by approximately 365,000 and 381,000, respectively, over the year ended December 31, 2017, representing growth of 25% and 27%, respectively.
All of our product revenue in the years ended December 31, 2018 and 2017 was derived from product sales of 1-gram and 0.5-gram size capsules of Vascepa, net of allowances, discounts, incentives, rebates, chargebacks and returns. The FDA-approved dosing for Vascepa continues to be 4 grams per day and, as expected, the majority of new and existing patients taking Vascepa continue to be prescribed the 1-gram size Vascepa capsules. Timing of shipments to wholesalers, as used for revenue recognition, and timing of prescriptions as estimated by third-party sources such as Symphony Health and IQVIA may differ from period to period.
During the years ended December 31, 2018 and 2017, our net product revenue included adjustment for co-pay mitigation rebates provided by us to commercially insured patients. Such rebates are intended to offset the differential for patients of Vascepa not covered by commercial insurers at the time of launch on Tier 2 for formulary purposes, resulting in higher co-pay amounts for such patients. Our cost for these co-payment mitigation rebates during the years ended December 31, 2018 and 2017 was up to $70 per 30-day prescription filled and, beginning in March 2017, included up to $140 per 90-day prescription filled. Since launch, certain third-party payors have added Vascepa to their Tier 2 coverage, which results in lower co-payments for patients covered by these third-party payors. In connection with such Tier 2 coverage, we have agreed to pay customary rebates to these third-party payors on the resale of Vascepa to patients covered by these third-party payors.
As is typical for the pharmaceutical industry, the majority of Vascepa sales are to major commercial wholesalers which then resell Vascepa to retail pharmacies.
78
Licensing Revenue. Licensing revenue during the years ended December 31, 2018 and 2017 was $0.8 million and $1.3 million, respectively, a decrease of $0.4 million, or 34%. Licensing revenue relates to the recognition of amounts received in connection with a Vascepa licensing agreement for the China Territory, specifically a $15.0 million up-front payment received in February 2015 and a $1.0 million milestone payment achieved in March 2016, as well as recognition of amounts received in connection with a Vascepa licensing agreement for Canada, specifically a $5.0 million up-front payment which was received upon closing of the agreement in September 2017 and a $2.5 million milestone payment that was received following achievement of the REDUCE-IT trial primary endpoint in September 2018. The up-front and milestone payments are being recognized over the estimated period in which we are required to provide regulatory and development support pursuant to the agreements. The amount of licensing revenue is expected to vary from period to period based on timing of milestones achieved and changes in estimates of the timing and level of support required. We do not anticipate significant revenues from international sources in 2019.
Cost of Goods Sold. Cost of goods sold during the years ended December 31, 2018 and 2017 was $54.5 million and $45.0 million, respectively, an increase of $9.6 million, or 21%. Cost of goods sold includes the cost of API for Vascepa on which revenue was recognized during the period, as well as the associated costs for encapsulation, packaging, shipment, supply management, insurance and quality assurance. The cost of the API included in cost of goods sold reflects the average cost of API included in inventory. This average cost reflects the actual purchase price of Vascepa API.
The API included in the calculation of the average cost of goods sold during the years ended December 31, 2018 and 2017 was sourced from multiple API suppliers. These suppliers compete with each other based on cost, consistent quality, capacity, timely delivery and other factors. In the future, we may see the average cost of supply change based on numerous potential factors including increased volume purchases, continued improvement in manufacturing efficiency, the mix of purchases made among suppliers, currency exchange rates and other factors. We currently anticipate API average cost in 2019 to be similar to or modestly lower than 2018. The average cost may be variable from period to period depending upon the timing and quantity of API purchased from each supplier.
Our gross margin on product sales for the years ended December 31, 2018 and 2017 was 76% and 75%, respectively.
Selling, General and Administrative Expense. Selling, general and administrative expense for the years ended December 31, 2018 and 2017 was $227.0 million and $134.5 million, respectively, an increase of $92.4 million, or 69%. Selling, general and administrative expenses for the years ended December 31, 2018 and 2017 are summarized in the table below:
|
|
|
Year Ended December 31, |
|
|||||
|
In thousands |
|
2018 |
|
|
2017 |
|
||
|
Selling, general and administrative expense (1) |
|
$ |
164,267 |
|
|
$ |
100,204 |
|
|
Co-promotion fees (2) |
|
|
46,821 |
|
|
|
22,507 |
|
|
Non-cash stock-based compensation expense (3) |
|
|
15,908 |
|
|
|
11,838 |
|
|
Total selling, general and administrative expense |
|
$ |
226,996 |
|
|
$ |
134,549 |
|
|
(1) |
Selling, general and administrative expense, excluding co-promotion fees and non-cash compensation charges for stock compensation, for the years ended December 31, 2018 and 2017 was $164.3 million and $100.2 million, respectively, an increase of $64.1 million, or 64%. This increase is due primarily to increased promotional activities, including commercial spend in preparation for successful REDUCE-IT results (announced on September 24, 2018) as well as costs for sales force expansion and other increased promotional activities following positive REDUCE-IT results. Incurred in 2018 and not incurred in 2017 were costs for direct to consumer activities of approximately $28 million, as well as payment of $2.0 million made in connection with the settlement agreement reached with Teva Pharmaceuticals USA, Inc. in May 2018. |
|
(2) |
Co-promotion fees payable to Kowa Pharmaceuticals America, Inc. were $46.8 million and $22.5 million in the years ended December 31, 2018 and 2017, respectively, an increase of $24.3 million, or 108%. The increase is due primarily to an accrual for co-promotion tail payments of $16.4 million in 2018 as well as an increase in gross margin on product sales, upon which the co-promotion fees are calculated for the twelve months ended December 31, 2018 compared to the same period in 2017. |
|
(3) |
Non-cash stock-based compensation expense for the years ended December 31, 2018 and 2017 was $15.9 million and $11.8 million, respectively, an increase of $4.1 million, or 34%. Non-cash stock-based compensation expense represents the estimated costs associated with equity awards issued to internal staff supporting our selling, general and administrative functions. The increase is due primarily to the determination in 2018 that certain performance awards are probable to be achieved following positive REDUCE-IT results, as well as an increase in the number of employees receiving equity awards as a result of the growth of our sales force and an increase in the underlying fair value of the equity awards resulting from the increase in the price of our stock. |
We anticipate our selling, general and administrative expenses to increase in 2019 as a result of increased promotional activities pertaining to Vascepa pursuant to positive REDUCE-IT results, including increasing the size of our sales force from approximately
79
170 sales professionals, including sales representatives and their managers, prior to the REDUCE-IT topline results announcement in September 2018, to approximately 440 sales professionals, including approximately 400 sales presentatives, to begin 2019, partially offset by the elimination of ongoing expense from co-promotion fees under the co-promotion agreement which, by design, ended on December 31, 2018. We anticipate selling, general and administrative expenses to increase further following label expansion of Vascepa by the FDA, subject to FDA review, including an anticipated increase in direct to consumer promotion.
Research and Development Expense. Research and development expense for the years ended December 31, 2018 and 2017 was $55.9 million and $47.2 million, respectively, an increase of $8.7 million, or 19%. Research and development expenses for the years ended December 31, 2018 and 2017 are summarized in the table below:
|
|
|
Year Ended December 31, |
|
|||||
|
In thousands |
|
2018 |
|
|
2017 |
|
||
|
REDUCE-IT study (1) |
|
$ |
38,098 |
|
|
$ |
34,886 |
|
|
Regulatory filing fees and expenses (2) |
|
|
1,052 |
|
|
|
1,011 |
|
|
Internal staffing, overhead and other (3) |
|
|
13,852 |
|
|
|
9,139 |
|
|
Research and development expense, excluding non-cash expense |
|
|
53,002 |
|
|
|
45,036 |
|
|
Non-cash stock-based compensation expense (4) |
|
|
2,898 |
|
|
|
2,122 |
|
|
Total research and development expense |
|
$ |
55,900 |
|
|
$ |
47,158 |
|
The increase in research and development expenses for the year ended December 31, 2018, as compared to the prior year period, is primarily due to timing of REDUCE-IT and related costs.
|
(1) |
In September 2018, we announced landmark positive topline results of the REDUCE-IT cardiovascular outcomes trial. The REDUCE-IT study met its primary endpoint demonstrating an approximately 25% relative risk reduction in composite of major adverse cardiovascular events with high statistical significance. This result was supported by robust demonstrations of efficacy across multiple secondary endpoints. The REDUCE-IT study results were further presented at the 2018 Scientific Sessions of the American Heart Association (AHA) on November 10, 2018 and concurrently published in The New England Journal of Medicine. We managed the study through a contract research organization (CRO) through which all costs for the conduct of this outcomes study were incurred with the exception of costs for clinical trial material (CTM) and costs for internal management. We expense costs for CTM when allocated to clinical research. For the years ended December 31, 2018 and 2017, we incurred expenses through our CRO in connection with this trial of approximately $29.1 million and $29.9 million, respectively. Inclusive of CTM costs, the combined CRO and CTM costs during the years ended December 31, 2018 and 2017 for REDUCE-IT were approximately $38.1 million and $34.9 million, respectively. Our internal personnel are responsible for managing multiple projects and their costs are not specifically allocated to REDUCE-IT or any other individual project. The increase in expenses in 2018 as compared to 2017 is primarily due to costs associated with last patient study visits and data collection and analysis related to the REDUCE-IT study. |
|
(2) |
The regulatory filing fees in each of the years ended December 31, 2018 and 2017 included annual FDA fees for maintaining manufacturing sites. Such fees primarily represent fees for qualification of new suppliers, including increasing capacity capabilities, and fees for sites used for the manufacture of product used in the REDUCE-IT clinical outcomes study. |
|
(3) |
Internal staffing, overhead and other research and development expenses primarily relate to the costs of our personnel employed to manage research, development and regulatory affairs activities and related overhead costs including consulting and other professional fees that are not allocated to specific projects. Also included are costs related to qualifying suppliers. These costs increased in 2018 compared to 2017 in support of publishing results of the REDUCE-IT study and preparing potential regulatory filings based on the results of the study. Other research and development expenses for the year ended December 31, 2018 include a non-refundable, non-creditable upfront payment of approximately $2.7 million related to our strategic collaboration with Mochida Pharmaceutical Co., Ltd. |
|
(4) |
Non-cash stock-based compensation expense represents the estimated costs associated with equity awards issued to internal staff supporting our research and development and regulatory functions. |
We anticipate our research and development expenses to be modestly lower in 2019 compared to 2018 as the REDUCE-IT trial is complete. Research and development expenses in 2019, while lower than in 2018, are anticipated to remain relatively high to support the sNDA submission, to support multiple potential additional publications of REDUCE-IT results, to support partners with respect to international submissions for Vascepa and to evaluate future product opportunities on our own and in collaboration with our development partner, Mochida.
80
Interest Expense, net. Net interest expense for the years ended December 31, 2018 and 2017 was $7.8 million and $9.3 million, respectively, a decrease of $1.5 million, or 9%. Net interest expense for the years ended December 31, 2018 and 2017 is summarized in the table below:
|
|
|
Year ended December 31, |
|
|||||
|
In thousands |
|
2018 |
|
|
2017 |
|
||
|
Exchangeable senior notes (1): |
|
|
|
|
|
|
|
|
|
Amortization of debt discounts |
|
$ |
186 |
|
|
$ |
200 |
|
|
Contractual coupon interest |
|
|
881 |
|
|
|
1,004 |
|
|
Total exchangeable senior notes interest expense |
|
|
1,067 |
|
|
|
1,204 |
|
|
Long-term debt from royalty-bearing instrument (2): |
|
|
|
|
|
|
|
|
|
Cash interest |
|
|
5,646 |
|
|
|
6,425 |
|
|
Non-cash interest |
|
|
1,997 |
|
|
|
2,132 |
|
|
Total long-term debt from royalty-bearing instrument interest expense |
|
|
7,643 |
|
|
|
8,557 |
|
|
Other interest expense |
|
|
162 |
|
|
|
5 |
|
|
Total interest expense |
|
|
8,872 |
|
|
|
9,766 |
|
|
Interest income (3) |
|
|
(1,074 |
) |
|
|
(429 |
) |
|
Total interest expense, net |
|
$ |
7,798 |
|
|
$ |
9,337 |
|
|
(1) |
Cash and non-cash interest expense related to the exchangeable senior notes, which were fully exchanged and retired for equity in November 2018, for the years ended December 31, 2018 and 2017 was $1.1 million and $1.2 million, respectively. |
|
(2) |
Cash and non-cash interest expense related to the December 2012 royalty-bearing instrument for the years ended December 31, 2018 and 2017 was $7.6 million and $8.6 million, respectively. These amounts reflect the fact that our Vascepa net revenue levels have not been, and during these years were not assumed to be, high enough to support repayment in accordance with the contractual repayment schedule without the optional reduction which is allowed to be elected by us if the threshold revenue levels are not achieved. To date, our revenues have been below the contractual threshold amount each quarter such that each payment reflects the calculated optional reduction amount as opposed to the contractual threshold payments for each quarterly period. |
|
(3) |
Interest income for the years ended December 31, 2018 and 2017 was $1.1 million and $0.4 million, respectively. Interest income represents income earned on cash balances. |
Other (Expense) Income, net. Other (expense) income, net, for the year ended December 31, 2018 and 2017 was expense of $0.3 million and income of $0.1 million, respectively. Other (expense) income, net, in the years ended December 31, 2018 and 2017 primarily consists of gains and losses on foreign exchange transactions.
Provision for Income Taxes. Provision for income taxes for the year ended December 31, 2018 and 2017 was $0.1 million and $13.0 million, respectively. The 2018 provision is the result of losses generated by our U.S. and non-U.S. operations for which no tax benefit has been recognized based on our position that deferred tax benefits are not more likely than not to be realized based on available evidence. The 2017 provision recorded related entirely to the U.S. subsidiary operations. At the date of enactment of the Tax Cuts and Jobs Act, we had net deferred tax assets for the excess of the net tax value over the book basis of our U.S. assets and liabilities which will generate future tax deductions in excess of book expense. As a result of the Tax Cuts and Jobs Act, future tax deductions will result in a decreased reduction in tax expense. Consequently, we reduced the amount of the U.S. subsidiary’s net deferred tax assets as of the date of enactment and recorded a non-cash charge of $2.4 million in the provision for income taxes for the year ended December 31, 2017 due to the decrease in the U.S. corporate tax rate from 34% to 21%. In addition, based on our evaluation of the available evidence, we recognized non-cash tax expense during the year ended December 31, 2017 of $8.7 million related to the recording of additional valuation allowance to reduce the deferred tax assets on the balance sheet to zero as we concluded that it is not more likely than not that certain of the deferred tax benefits resulting from the deferred tax assets generated from the U.S. subsidiary operations will be realized.
The provisions for income taxes for the years ended December 31, 2018 and 2017 include excess tax benefits of $7.7 million and $1.3 million, respectively, arising from share-based payments as a result of adopting ASU No. 2016-09, Compensation—Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting, which requires that excess tax benefits and deficiencies that arise upon vesting or exercise of share-based payments be recognized as an income tax benefit and expense in the income statement.
Comparison of Fiscal Years Ended December 31, 2017 and December 31, 2016
Product Revenue, net. We recorded net product revenue of $179.8 million and $129.0 million during the years ended December 31, 2017 and 2016, respectively, an increase of $50.9 million, or 39%. This increase in revenue was driven primarily by an
81
increase in estimated normalized total Vascepa prescriptions. Based on data provided by Symphony Health and IQVIA (formerly QuintilesIMS), estimated normalized total Vascepa prescriptions increased by approximately 441,000 and 425,000, respectively, over the year ended December 31, 2016, representing growth of 45% and 42%, respectively. During 2017, overall wholesaler inventory levels decreased from year-end 2016 levels calculated based on estimated days of Vascepa sales on hand. We believe that changes in channel inventory at these independent wholesalers and retail pharmacies are common and are impacted by numerous factors, including holiday timing and recent order trends. We also believe, based on information available to us, that channel inventory levels at the end of both 2017 and 2016 were within ordinary ranges.
All of our product revenue in the years ended December 31, 2017 and 2016 was derived from product sales of 1-gram and 0.5-gram size capsules of Vascepa, net of allowances, discounts, incentives, rebates, chargebacks and returns. The FDA-approved dosing for Vascepa continues to be 4 grams per day and, as expected, the majority of new and existing patients taking Vascepa continue to be prescribed the 1-gram size Vascepa capsules. Timing of shipments to wholesalers, as used for revenue recognition, and timing of prescriptions as estimated by third-party sources such as Symphony Health and IQVIA (formerly QuintilesIMS) may differ from period to period.
During the years ended December 31, 2017 and 2016, our net product revenue included an adjustment for co-pay mitigation rebates provided by us to commercially insured patients. Such rebates are intended to offset the differential for patients of Vascepa not covered by commercial insurers at the time of launch on Tier 2 for formulary purposes, resulting in higher co-pay amounts for such patients. Our cost for these co-payment mitigation rebates during the years ended December 31, 2017 and 2016 was up to $70 per 30-day prescription filled and, beginning in March 2017, included up to $140 per 90-day prescription filled. Since launch, certain third-party payors have added Vascepa to their Tier 2 coverage, which results in lower co-payments for patients covered by these third-party payors. In connection with such Tier 2 coverage, we have agreed to pay customary rebates to these third-party payors on the resale of Vascepa to patients covered by these third-party payors.
As is typical for the pharmaceutical industry, the majority of Vascepa sales are to major commercial wholesalers which then resell Vascepa to retail pharmacies.
Licensing Revenue. Licensing revenue during the years ended December 31, 2017 and 2016 was $1.3 million and $1.1 million, respectively, an increase of $0.2 million, or 14%. Licensing revenue relates to the amortization of a $15.0 million up-front payment received in February 2015 and a $1.0 million milestone payment achieved in March 2016, both associated with a Vascepa licensing agreement for the China Territory. Licensing revenue also includes amortization of a $5.0 million up-front amount associated with a Vascepa licensing agreement for Canada, which was reached in September 2017. The up-front and milestone payments are being recognized over the estimated period in which we are required to provide regulatory and development support and clinical and commercial supply under the agreements. The amount of licensing revenue recorded may be variable from period to period based on timing of milestones achieved and changes in estimates of the timing and level of support required.
Cost of Goods Sold. Cost of goods sold during the years ended December 31, 2017 and 2016 was $45.0 million and $34.4 million, respectively, an increase of $10.6 million, or 31%. Cost of goods sold includes the cost of API for Vascepa on which revenue was recognized during the period, as well as the associated costs for encapsulation, packaging, shipment, supply management, insurance and quality assurance. The cost of the API included in cost of goods sold reflects the average cost of API included in inventory. This average cost reflects the actual purchase price of Vascepa API.
The API included in the calculation of the average cost of goods sold during the years ended December 31, 2017 and 2016 was sourced from three API suppliers. These suppliers compete with each other based on cost, consistent quality, capacity, timely delivery and other factors. In the future, we may see the average cost of supply change based on numerous potential factors including increased volume purchases, continued improvement in manufacturing efficiency, the mix of purchases made among suppliers, currency exchange rates and other factors. We currently anticipate API average cost in 2018 to be similar to 2017. The average cost may be variable from period to period depending upon the timing and quantity of API purchased from each supplier.
Our gross margin on product sales for the years ended December 31, 2017 and 2016 was 75% and 73%, respectively. This improvement was primarily driven by lower unit cost API purchases.
82
Selling, General and Administrative Expense. Selling, general and administrative expense for the years ended December 31, 2017 and 2016 was $134.5 million and $111.4 million, respectively, an increase of $23.2 million, or 21%. Selling, general and administrative expenses for the years ended December 31, 2017 and 2016 are summarized in the table below:
|
|
|
Year Ended December 31, |
|
|||||
|
In thousands |
|
2017 |
|
|
2016 |
|
||
|
Selling, general and administrative expense (1) |
|
$ |
100,204 |
|
|
$ |
82,042 |
|
|
Co-promotion fees (2) |
|
|
22,507 |
|
|
|
17,969 |
|
|
Non-cash stock-based compensation expense (3) |
|
|
11,838 |
|
|
|
11,361 |
|
|
Total selling, general and administrative expense |
|
$ |
134,549 |
|
|
$ |
111,372 |
|
|
(1) |
Selling, general and administrative expense, excluding co-promotion fees and non-cash compensation charges for stock compensation, for the years ended December 31, 2017 and 2016 was $100.2 million and $82.0 million, respectively, an increase of $18.2 million, or 22%. This increase is due primarily to increased promotional activities, including commercial spend for anticipated expansion following successful REDUCE-IT results, and increased legal costs, which are subject to quarterly variability. |
|
(2) |
Co-promotion fees payable to Kowa Pharmaceuticals America, Inc. were $22.5 million and $18.0 million in the years ended December 31, 2017 and 2016, respectively, an increase of $4.5 million, or 25%. The increase is due primarily to an increase in gross margin on product sales for the year ended December 31, 2017 compared to the same period in 2016, offset as a result of amended contract terms in 2017 related to the percentage of aggregate Vascepa gross margin earned by Kowa Pharmaceuticals America, Inc. and certain other refinements. |
|
(3) |
Non-cash stock-based compensation expense for the years ended December 31, 2017 and 2016 was $11.8 million and $11.4 million, respectively, an increase of $0.5 million, or 4%. |
Research and Development Expense. Research and development expense for the years ended December 31, 2017 and 2016 was $47.2 million and $50.0 million, respectively, a decrease of $2.8 million, or 6%. Research and development expenses for the years ended December 31, 2017 and 2016 are summarized in the table below:
|
|
|
Year Ended December 31, |
|
|||||
|
In thousands |
|
2017 |
|
|
2016 |
|
||
|
REDUCE-IT study (1) |
|
$ |
34,886 |
|
|
$ |
36,989 |
|
|
Regulatory filing fees and expenses (2) |
|
|
1,011 |
|
|
|
1,735 |
|
|
Internal staffing, overhead and other (3) |
|
|
9,139 |
|
|
|
8,999 |
|
|
Research and development expense, excluding non-cash expense |
|
|
45,036 |
|
|
|
47,723 |
|
|
Non-cash stock-based compensation expense (4) |
|
|
2,122 |
|
|
|
2,252 |
|
|
Total research and development expense |
|
$ |
47,158 |
|
|
$ |
49,975 |
|
The decrease in research and development expenses for the year ended December 31, 2017, as compared to the prior year period, is primarily due to timing of REDUCE-IT and related costs.
|
(1) |
In December 2011, we announced commencement of patient dosing in our cardiovascular outcomes study of Vascepa, titled REDUCE-IT, which is designed to evaluate the efficacy of Vascepa, including its impact on triglyceride lowering and its other clinical effects, in reducing major adverse cardiovascular events compared to placebo in patients who despite statin therapy have risk factors for cardiovascular disease, including elevated triglyceride levels. In 2016, we completed patient enrollment and randomization of 8,179 individual patients into the REDUCE-IT study, exceeding the 8,000 patients targeted for the trial. We manage the study through a contract research organization (CRO) through which all costs for this outcomes study are incurred with the exception of costs for clinical trial material (CTM) and costs for internal management. Our internal personnel are responsible for managing multiple projects and their costs are not specifically allocated to REDUCE-IT or any other individual project. For the years ended December 31, 2017 and 2016, we incurred expenses through our CRO in connection with this trial of approximately $29.9 million and $28.8 million, respectively. Inclusive of CTM costs, the combined CRO and CTM costs during the years ended December 31, 2017 and 2016 for REDUCE-IT were approximately $34.9 million and $37.0 million, respectively. The decrease in expenses in 2017 as compared to 2016 is primarily the result of timing variability for REDUCE-IT costs. We expense costs for CTM when allocated to clinical research. |
|
(2) |
The regulatory filing fees in each of the years ended December 31, 2017 and 2016 included annual FDA fees for maintaining manufacturing sites. Such fees primarily represent fees for qualification of new suppliers, including increasing capacity capabilities, and fees for sites used for the manufacture of product used in the REDUCE-IT clinical outcomes study. |
|
(3) |
Internal staffing, overhead and other research and development expenses primarily relate to the costs of our personnel employed to manage research, development and regulatory affairs activities and related overhead costs including consulting and other professional fees that are not allocated to specific projects. Also included are costs related to qualifying suppliers. |
83
|
(4) |
Non-cash stock-based compensation expense represents the costs associated with equity awards issued to internal staff supporting our research and development and regulatory functions. |
Gain on Change in Fair Value of Derivative Liabilities. Gain on change in fair value of derivative liabilities for the year ended December 31, 2017 was nil versus a gain of $8.2 million in the prior year period. Gain on change in fair value of derivative liabilities for the years ended December 31, 2017 and 2016 is comprised of (i) the change in fair value of the derivative liability related to the change in control provision associated with the December 2012 royalty-bearing instrument, and (ii) the change in fair value of the derivative liabilities related to the change in control provisions associated with the May 2014 and November 2015 exchangeable senior notes.
Our December 2012 royalty-bearing instrument financing arrangement with CPPIB Credit Europe S.à r.l., or CPPIB, as successor in interest to BioPharma Secured Debt Fund II Holdings Cayman LP, contains a redemption feature whereby, upon a change of control, we would be required to repay $150.0 million, less any previously repaid amount, which net remaining unpaid amount as of December 31, 2017 was $109.1 million. Unless this early redemption feature is triggered, the remaining amount, without additional interest accumulation, is anticipated to be paid based on the royalty provisions of the agreement. The fair value of the derivative liability is recalculated at each reporting period using a probability-weighted model incorporating management estimates for potential change in control, and by determining the fair value of the debt with and without the change in control provision included. The difference between the two fair values of the debt was determined to be the fair value of the embedded derivative. No gain or loss on change in fair value of derivative liability was recognized for the year ended December 31, 2017 as the fair value of the derivative was determined to be nil based on underlying assumptions as of both December 31, 2016 and December 31, 2017. As of December 31, 2015, the fair value of the derivative was determined to be $5.5 million, and as of December 31, 2016, the fair value of the derivative was determined to be nil. As such, we recognized a $5.5 million gain on change in fair value of derivative liability for the year ended December 31, 2016.
Our 3.5% May 2014 exchangeable senior notes due 2032, or 2014 Notes, contained a redemption feature whereby, upon occurrence of a change in control, we would have been required to repurchase the notes. The fair value of the embedded derivative was calculated using a probability-weighted model incorporating management estimates of the probability of a change in control occurring, and by determining the fair value of the debt with and without the change in control provision included. The difference between the two was determined to be the fair value of the embedded derivative. In September 2016, we exercised our optional exchange rights upon satisfaction of specified equity conditions set forth in the 2014 Notes indenture to mandatorily exchange the 2014 Notes into ADSs (see Note 7—Debt). As such, the related derivative liability was derecognized at that time and we recognized a $2.1 million gain on change in fair value of derivative liability for the year ended December 31, 2016. There was no such change in fair value of derivative liability for the year ended December 31, 2017.
Our 3.5% November 2015 exchangeable senior notes due 2032, or 2015 Notes, contained the same redemption feature as the 2014 Notes and the related derivative liability was calculated utilizing the same methodology. In September 2016, we exercised our optional exchange rights upon satisfaction of specified equity conditions consistent with the terms of the 2015 Notes to mandatorily exchange the 2015 Notes into ADSs (see Note 7—Debt). As such, the related derivative liability was derecognized at that time and we recognized a $0.6 million gain on change in fair value of derivative liability for the year ended December 31, 2016. There was no such change in fair value of derivative liability for the year ended December 31, 2017.
The change in fair value of the derivative liability related to the royalty-bearing instrument is largely related to our projections for future estimated revenues, management estimates of the probability of a change in control occurring, and the terms of debt issues of similar companies. The change in fair value of the derivative liabilities related to the 2014 Notes and 2015 Notes was a result of the exchange of the related debt hosts for the year ended December 31, 2016. Any changes in the assumptions used to value the derivative liabilities could result in a material change to the carrying value of such liabilities.
Interest Expense, net. Net interest expense for the years ended December 31, 2017 and 2016 was $9.3 million and $18.4 million, respectively, a decrease of $9.1 million, or 48%. Net interest expense for the years ended December 31, 2017 and 2016 is summarized in the table below:
84
|
|
|
Year ended December 31, |
|
|||||
|
In thousands |
|
2017 |
|
|
2016 |
|
||
|
Exchangeable senior notes (1): |
|
|
|
|
|
|
|
|
|
Amortization of debt discounts |
|
$ |
200 |
|
|
$ |
5,703 |
|
|
Contractual coupon interest |
|
|
1,004 |
|
|
|
4,151 |
|
|
Total exchangeable senior notes interest expense |
|
|
1,204 |
|
|
|
9,854 |
|
|
Long-term debt from royalty-bearing instrument (2): |
|
|
|
|
|
|
|
|
|
Cash interest |
|
|
6,425 |
|
|
|
6,727 |
|
|
Non-cash interest |
|
|
2,132 |
|
|
|
2,081 |
|
|
Total long-term debt from royalty-bearing instrument interest expense |
|
|
8,557 |
|
|
|
8,808 |
|
|
Other interest expense |
|
|
5 |
|
|
|
15 |
|
|
Total interest expense |
|
|
9,766 |
|
|
|
18,677 |
|
|
Interest income (3) |
|
|
(429 |
) |
|
|
(234 |
) |
|
Total interest expense, net |
|
$ |
9,337 |
|
|
$ |
18,443 |
|
|
(1) |
Cash and non-cash interest expense related to the exchangeable senior notes for the years ended December 31, 2017 and 2016 was $1.2 million and $9.9 million, respectively. The decrease in cash and non-cash interest expense is the result of the decrease in principal amount of exchangeable senior notes from $165.1 million outstanding during the majority of 2016 to $30.0 million outstanding during 2017. |
|
(2) |
Cash and non-cash interest expense related to the December 2012 royalty-bearing instrument for the years ended December 31, 2017 and 2016 was $8.6 million and $8.8 million, respectively. These amounts reflect the fact that our Vascepa net revenue levels have not been, and during these years were not assumed to be, high enough to support repayment in accordance with the contractual repayment schedule without the optional reduction which is allowed to be elected by us if the threshold revenue levels are not achieved. To date, our revenues have been below the contractual threshold amount each quarter such that each payment reflects the calculated optional reduction amount as opposed to the contractual threshold payments for each quarterly period. |
|
(3) |
Interest income for the years ended December 31, 2017 and 2016 was $0.4 million and $0.2 million, respectively. Interest income represents income earned on cash balances. |
Other Expense (Income), net. Other expense (income), net, for the year ended December 31, 2017 and 2016 was income of $0.1 million and expense of $0.5 million, respectively. Other expense (income), net, in the years ended December 31, 2017 and 2016 primarily consists of gains and losses on foreign exchange transactions.
Provision for Income Taxes. Provision for income taxes for the year ended December 31, 2017 and 2016 was $13.0 million and $10.0 million, respectively. The provisions recorded relate entirely to the U.S. subsidiary operations. At the date of enactment of the Tax Cuts and Jobs Act, we had net deferred tax assets for the excess of the net tax value over the book basis of our U.S. assets and liabilities which will generate future tax deductions in excess of book expense. As a result of the Tax Cuts and Jobs Act, future tax deductions will result in a decreased reduction in tax expense. Consequently, we reduced the amount of the U.S. subsidiary’s net deferred tax assets as of the date of enactment and recorded a non-cash charge of $2.4 million in the provision for income taxes for the year ended December 31, 2017 due to the decrease in the U.S. corporate tax rate from 34% to 21%. In addition, based on our evaluation of the available evidence, we recognized non-cash tax expense during the year ended December 31, 2017 of $8.7 million related to the recording of additional valuation allowance to reduce the deferred tax assets on the balance sheet to zero as we concluded that it is not more likely than not that certain of the deferred tax benefits resulting from the deferred tax assets generated from the U.S. subsidiary operations will be realized. The provisions for income taxes for the years ended December 31, 2017 and 2016 include $1.3 million of excess tax benefits and $0.4 million of excess tax deficiencies, respectively, arising from share-based payments as a result of adopting ASU No. 2016-09, Compensation—Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting, which requires that excess tax benefits and deficiencies that arise upon vesting or exercise of share-based payments be recognized as an income tax benefit and expense in the income statement.
Liquidity and Capital Resources
Our sources of liquidity as of December 31, 2018 include cash and cash equivalents of $249.2 million. In February 2018, we completed a public offering of 19,178,082 ADSs and, in March 2018, we issued an additional 1,438,356 ADSs upon the underwriter’s partial exercise of a 30-day option to purchase additional shares. The underwriter purchased the ADSs from us at a price of $3.41 per ADS after commission, resulting in net proceeds to us of approximately $70.0 million, after deducting customary commissions and offering expenses. In November 2018, we completed a public offering of 11,111,112 ADSs. The underwriters purchased the ADSs from us at a price of $17.575 per ADS after commission, resulting in net proceeds to us of approximately $70.0 million, after
85
deducting customary commissions and offering expenses. Our projected uses of cash include expansion of our sales force and initiatives for medical education and market awareness following successful REDUCE-IT results, increasing inventory purchases, and general corporate and working capital purposes. Our cash flows from operating, investing and financing activities, as reflected in the consolidated statements of cash flows, are summarized in the following table:
|
|
|
Year Ended December 31, |
|
|||||||||
|
In millions |
|
2018 |
|
|
2017 |
|
|
2016 |
|
|||
|
Cash (used in) provided by: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating activities |
|
$ |
(94.7 |
) |
|
$ |
(32.8 |
) |
|
$ |
(71.8 |
) |
|
Investing activities |
|
|
(0.1 |
) |
|
|
— |
|
|
|
— |
|
|
Financing activities |
|
|
271.3 |
|
|
|
8.2 |
|
|
|
63.1 |
|
|
Increase (decrease) in cash and cash equivalents |
|
$ |
176.5 |
|
|
$ |
(24.6 |
) |
|
$ |
(8.7 |
) |
Net cash used in operating activities during 2018 compared to 2017 increased primarily as a result of commercial and R&D activities associated with preparation for completion of the REDUCE-IT study, including payment for increased levels of Vascepa inventory, as well as costs to expand commercial activities and publish topline results of the REDUCE-IT study which were reported in September 2018. These increases were partially offset by higher collections from product sales.
In December 2012, we entered into a financing agreement with BioPharma. Under this agreement, we granted to BioPharma a security interest in future receivables and all related rights to Vascepa, in exchange for $100.0 million received at the closing of the agreement which closing occurred in December 2012. In December 2017, BioPharma assigned all rights under this agreement to CPPIB. We have agreed to repay up to $150.0 million of future revenue and receivables. As of December 31, 2018, the net remaining amount to be repaid to CPPIB is $88.6 million, which will be repaid in quarterly installments calculated as 10% of quarterly Vascepa net revenues. We can prepay the net remaining amount at any time.
As of December 31, 2018, we have no exchangeable notes or term debt outstanding since, in October 2018, we exercised our optional exchange rights upon satisfaction of specified equity conditions set forth in the 3.5% exchangeable senior notes due 2047, or the 2017 Notes, to mandatorily exchange the entirety of the $30.0 million in aggregate principal amount outstanding into ADSs. This resulted in elimination of the debt and issuance of 7,716,046 ADSs. The 2017 Notes were issued and sold in January 2017 when we, through our wholly-owned subsidiary Corsicanto II DAC, or Corsicanto II, a private designated activity company incorporated under the laws of Ireland, entered into separate, privately negotiated purchase agreements with certain unrelated investors. The net proceeds we received from the January 2017 offering were approximately $28.8 million, after deducting placement agent fees and estimated offering expenses. See Note 7—Debt in the Notes to Consolidated Financial Statements for further discussion.
As of December 31, 2018, we had cash and cash equivalents of $249.2 million, an increase of $175.6 million from December 31, 2017. The increase is primarily due to proceeds from the public offering financings completed in the first and fourth quarters of 2018 and accounts receivable collections, partially offset by net cash used in operating activities in support of the commercialization of Vascepa and funding of REDUCE-IT. As of December 31, 2018, we had accounts receivable, net, of $66.5 million and inventory, net, of $57.8 million. We have incurred annual operating losses since our inception and, as a result, we had an accumulated deficit of $1.4 billion as of December 31, 2018. We anticipate that quarterly net cash outflows in future periods will continue to be variable as a result of the timing of certain items, including our purchases of API and expanded Vascepa promotional activities resulting from positive REDUCE-IT results both before and after label change for Vascepa based on REDUCE-IT results, which label change is subject to FDA review and approval of our sNDA. Because levels of Vascepa revenues are difficult to predict, as is the timing of label expansion, we intend to purchase API during 2019 at a rate which, based on current levels of revenue growth, is higher than is required for 2019. We estimate the incremental cost of this anticipated inventory build to be between $50 million and $75 million in 2019. We believe that there is limited financial risk of over-purchasing Vascepa inventory as the product has demonstrated stability supporting approved commercial expiry dating through four years.
We believe that our cash and cash equivalents of $249.2 as of December 31, 2018, will be sufficient to fund our projected operations for at least twelve months and through the likely PDUFA date for approval of a supplemental new drug application (sNDA) by the FDA based on REDUCE-IT study results. Depending on the level of cash generated from operations, and depending in part on the timing and results of the FDA review of the sNDA and rate of prescription growth for Vascepa, additional capital may be required to support planned expansion of Vascepa promotion and potential Vascepa promotion beyond which we are currently executing. If additional capital is required and we are unable to obtain additional capital, we may be forced to delay, limit or eliminate certain promotional activities.
86
The following table summarizes our contractual obligations as of December 31, 2018 and the effects such obligations are expected to have on our liquidity and cash flows in future periods:
Payments Due by Period
|
In millions |
|
Total |
|
|
2019 |
|
|
2020 to 2021 |
|
|
2022 to 2023 |
|
|
After 2023 |
|
|||||
|
Contractual obligations: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase obligations (1) |
$ |
53.0 |
|
|
$ |
11.3 |
|
|
$ |
22.6 |
|
|
$ |
17.2 |
|
|
$ |
1.9 |
|
|
|
Operating lease obligations (2) |
|
0.2 |
|
|
|
0.2 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
Total contractual cash obligations |
$ |
53.2 |
|
|
$ |
11.5 |
|
|
$ |
22.6 |
|
|
$ |
17.2 |
|
|
$ |
1.9 |
|
|
|
(2) |
Represents operating lease costs, primarily consisting of leases for facilities in Dublin, Ireland and Bedminster, NJ, net of sublease rental income. |
As of December 31, 2018, we had certain marketing commitments, consisting of communication costs related to our direct-to-consumer activities, totaling approximately $1.2 million.
Under the terms of the agreement with CPPIB, as successor in interest to BioPharma, we agreed to repay up to $150.0 million of future revenue and receivables. As of December 31, 2018, the net remaining amount to be repaid is $88.6 million, which will be repaid in quarterly installments calculated as 10% of quarterly Vascepa net revenues. We can prepay the net remaining amount at any time.
Under the 2004 share repurchase agreement with Laxdale Limited, or Laxdale, upon approval of Vascepa by the FDA on July 26, 2012, we were required to make a milestone payment to Laxdale of £7.5 million. We made this payment in 2012 and capitalized this Laxdale milestone payment of $11.6 million as a component of other long-term assets. This long-term asset is being amortized over the estimated useful life of the intellectual property we acquired from Laxdale and we recognized amortization expense of $0.6 million in each of the years ended December 31, 2018 and 2017. Also under the Laxdale agreement, upon receipt of marketing approval in Europe for the first indication for Vascepa (or first indication of any product containing Amarin Neuroscience Limited intellectual property acquired from Laxdale in 2004), we must make an aggregate stock or cash payment to the former shareholders of Laxdale (at the sole option of each of the sellers) of £7.5 million (approximately $9.6 million as of December 31, 2018). Additionally, upon receipt of a marketing approval in the United States or Europe for a further indication of Vascepa (or further indication of any other product using Amarin Neuroscience Limited intellectual property), we must make an aggregate stock or cash payment (at the sole option of each of the sellers) of £5 million (approximately $6.4 million as of December 31, 2018) for each of the two potential market approvals (i.e., £10 million maximum, or approximately $12.7 million as of December 31, 2018).
We do not enter into financial instruments for trading or speculative purposes. As of December 31, 2018 and 2017, we had no outstanding forward exchange contracts.
Off-Balance Sheet Arrangements
We do not have any special purpose entities or other off-balance sheet arrangements.
We are exposed to market risks, which include changes in interest rates. We do not use derivative financial instruments in our investment portfolio, and other than in 2013, we enter into no foreign exchange contracts. Our investments meet high credit quality and diversification standards, as specified in our investment policy.
Foreign Currency Exchange Risk. Our results of operations and cash flows are subject to fluctuations due to changes in the Euro, Sterling and Yen. The majority of cash and cash equivalents and the majority of our vendor relationships are denominated in U.S. dollars. We therefore believe that the risk of a significant impact on our operating income from foreign currency fluctuations is not substantial. From time to time, we maintain a small amount of our cash and cash equivalents in Euro and Pound Sterling. We purchase a portion of our supply from Novasep based on a U.S. dollar to Euro exchange rate and as such, remain subject to currency fluctuation risk for such purchases.
87
Interest Rate Risk. We believe that we are not exposed to significant interest rate risk through market value fluctuations of balance sheet items (i.e., price risk) or through changes in interest income or expenses (i.e., re-financing or re-investment risk). Interest rate risk mainly arises through interest bearing liabilities and assets. We invest funds not needed for near-term operating expenses in diversified short-term investments, consisting primarily of investment grade securities. As of December 31, 2018, the fair value of our cash and cash equivalents maturing in one year or less was $249.2 million and represented 100% of our cash, cash equivalents and investment portfolio. A hypothetical 50 basis point change in interest rates would not result in a material decrease or increase in the fair value of our securities due to the general short-term nature of our investment portfolio.
Our consolidated financial statements are annexed to this report beginning on page F-1.
None.
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is (1) recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and (2) accumulated and communicated to our management, including our Principal Executive Officer and Principal Financial Officer, to allow timely decisions regarding required disclosure.
As of December 31, 2018 (the “Evaluation Date”), our management, with the participation of our Principal Executive Officer and Principal Financial Officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act). Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Our Principal Executive Officer and Principal Financial Officer have concluded based upon the evaluation described above that, as of the Evaluation Date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting for our company. Internal control over financial reporting is defined in Rules 13a-15(f) and 15(d)-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, our Principal Executive Officer and Principal Financial Officer and effected by our board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
|
|
• |
pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and disposition of our assets; |
|
|
• |
provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles; |
|
|
• |
provide reasonable assurance that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and |
|
|
• |
provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements. |
Because of inherent limitations, internal controls over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risks that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
Our management, including our Principal Executive Officer and Principal Financial Officer, has conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2018. In conducting this evaluation, we used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), in Internal Control-Integrated Framework (2013).
88
Based upon this evaluation and those criteria, management believes that, as of December 31, 2018, our internal controls over financial reporting were effective.
Ernst & Young LLP, our independent registered public accounting firm, has audited our consolidated financial statements and the effectiveness of our internal control over financial reporting as of December 31, 2018. This report appears below.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the fourth quarter of 2018 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
89
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors of Amarin Corporation plc
Opinion on Internal Control over Financial Reporting
We have audited Amarin Corporation plc’s internal control over financial reporting as of December 31, 2018, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Amarin Corporation plc (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2018, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2018 and 2017, the related consolidated statements of operations, stockholders’ deficit and cash flows for each of the three years in the period ended December 31, 2018 and the related notes and our report dated February 27, 2019 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Iselin, New Jersey
February 27, 2019
90
Entry into Rule 10b5-1 Trading Plans
Our policy governing transactions in our securities by our directors, officers and employees permits our officers, directors and certain other persons to enter into trading plans complying with Rule 10b5-1 under the Securities Exchange Act of 1934, as amended. Consistent with such regulation, our policy permits such plans to be entered into only when that person confirms they are not in possession of material non-public information. Our policy also requires a waiting period after a trading plan is created before shares can be traded under the plan. Our open trading windows are established in consultation with legal counsel. We have been from time to time advised that a number of our directors and employees, including members of our senior management team, and investment funds associated with such persons, have entered into trading plans in accordance with Rule 10b5-1 and our policy governing transactions in our securities. It is not our policy to publicly disclose the terms of these private trading plans. We undertake no obligation to update or revise the information provided herein, including for revision or termination of an established trading plan.
91
The information required by this item will be contained in our definitive proxy statement, which will be filed with the SEC in connection with our 2019 Annual General Meeting of Shareholders. Such information is incorporated herein by reference.
Code of Ethics
Our Board of Directors has adopted a code of business conduct and ethics that applies to our directors, officers and employees. There have been no material modifications to, or waivers from, the provisions of such code. This code is available on the corporate governance section of our website (which is a subsection of the investor relations section of our website) at the following address: www.amarincorp.com. Any waivers from or amendments to the code will be filed with the SEC on Form 8-K. You may also request a printed copy of the code, without charge, by writing to us at Amarin Pharma, Inc., 1430 Route 206, Bedminster, NJ 07921, Attention: Investor Relations.
The information required by this item will be contained in our definitive proxy statement, which will be filed with the SEC in connection with our 2019 Annual General Meeting of Shareholders. Such information is incorporated herein by reference.
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this item will be contained in our definitive proxy statement, which will be filed with the SEC in connection with our 2019 Annual General Meeting of Shareholders. Such information is incorporated herein by reference.
The information required by this item will be contained in our definitive proxy statement, which will be filed with the SEC in connection with our 2019 Annual General Meeting of Shareholders. Such information is incorporated herein by reference.
The information required by this item will be contained in our definitive proxy statement, which will be filed with the SEC in connection with our 2019 Annual General Meeting of Shareholders. Such information is incorporated herein by reference.
92
|
Exhibit |
|
|
|
Incorporated by Reference Herein |
||
|
Number |
|
Description |
|
Form |
|
Date |
|
|
|
|
|
|
|
|
|
3.1 |
|
|
Quarterly Report on Form 10-Q, File No. 0-21392, as Exhibit 3.1 |
|
August 8, 2013 |
|
|
|
|
|
|
|
|
|
|
4.1 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2011, File No. 0-21392, as Exhibit 4.1 |
|
February 29, 2012 |
|
|
|
|
|
|
|
|
|
|
4.2 |
|
|
Current Report on Form 8-K dated January 9, 2012, File No. 0-21392, as Exhibit 4.1 |
|
January 10, 2012 |
|
|
|
|
|
|
|
|
|
|
4.3 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2002, File No. 0-21392, as Exhibit 2.4 |
|
April 24, 2003 |
|
|
|
|
|
|
|
|
|
|
4.4 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2011, File No. 0-21392, as Exhibit 4.4 |
|
February 29, 2012 |
|
|
|
|
|
|
|
|
|
|
4.5 |
|
|
Current Report on Form 8-K dated March 5, 2015, File No. 0-21392, as Exhibit 4.1 |
|
March 11, 2015 |
|
|
|
|
|
|
|
|
|
|
4.6 |
|
|
Current Report on Form 8-K dated March 30, 2015, File No. 0-21392, as Exhibit 4.1 |
|
March 30, 2015 |
|
|
|
|
|
|
|
|
|
|
4.7 |
|
|
Current Report on Form 8-K dated March 30, 2015, File No. 0-21392, as Exhibit 4.2 |
|
March 30, 2015 |
|
|
|
|
|
|
|
|
|
|
10.1 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2006, File No. 0-21392, as Exhibit 4.17 |
|
March 5, 2007 |
|
|
|
|
|
|
|
|
|
|
10.2 |
|
|
Quarterly Report on Form 10-Q for the period ended June 30, 2011, File No. 0-21392, as Exhibit 10.4 |
|
August 9, 2011 |
|
|
|
|
|
|
|
|
|
|
10.3 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended June 30, 2012, File No. 0-21392, as Exhibit 10.1 |
|
August 8, 2008 |
|
|
|
|
|
|
|
|
|
|
10.4 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended June 30, 2012, File No. 0-21392, as Exhibit 10.2 |
|
August 8, 2008 |
|
93
|
Exhibit |
|
|
|
Incorporated by Reference Herein |
||
|
Number |
|
Description |
|
Form |
|
Date |
|
|
|
|
|
|
|
|
|
10.5 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2012, File No. 0-21392, as Exhibit 10.5 |
|
February 28, 2012 |
|
|
|
|
|
|
|
|
|
|
10.6 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended June 30, 2015, File No. 0-21392, as Exhibit 4.1 |
|
August 6, 2015 |
|
|
|
|
|
|
|
|
|
|
10.7 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended June 30, 2015, File No. 0-21392, as Exhibit 4.2 |
|
August 6, 2015 |
|
|
|
|
|
|
|
|
|
|
10.8 |
|
Amarin Corporation plc Management Incentive Compensation Plan* |
|
Annual Report on Form 10-K for the year ended December 31, 2010, File No. 0-21392, as Exhibit 10.44 |
|
March 16, 2011 |
|
|
|
|
|
|
|
|
|
10.9 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2011, File No. 0-21392, as Exhibit 10.3 |
|
February 29, 2012 |
|
|
|
|
|
|
|
|
|
|
10.10 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2011, File No. 0-21392, as Exhibit 10.4 |
|
February 29, 2012 |
|
|
|
|
|
|
|
|
|
|
10.11 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2011, File No. 0-21392, as Exhibit 10.5 |
|
February 29, 2012 |
|
|
|
|
|
|
|
|
|
|
10.12 |
|
Letter Agreement, dated November 15, 2010, between the Company and John F. Thero* |
|
Annual Report on Form 10-K for the year ended December 31, 2010, File No. 0-21392, as Exhibit 10.42 |
|
March 16, 2011 |
|
|
|
|
|
|
|
|
|
10.13 |
|
Letter Agreement with Joseph Kennedy, dated December 13, 2011* |
|
Current Report on Form 8-K dated December 23, 2011, File No. 0-21392, as Exhibit 10.5 |
|
December 23, 2011 |
|
|
|
|
|
|
|
|
|
10.14 |
|
|
Current Report on Form 8-K dated December 23, 2011, File No. 0-21392, as Exhibit 10.1 |
|
December 23, 2011 |
|
|
|
|
|
|
|
|
|
|
10.15 |
|
Letter Agreement with Steve Ketchum, dated February 8, 2012* |
|
Current Report on Form 8-K dated February 16, 2012, File No. 0-21392, as Exhibit 10.1 |
|
February 16, 2012 |
|
|
|
|
|
|
|
|
|
10.16 |
|
|
Current Report on Form 8-K dated January 8, 2014, File No. 0-21392, as Exhibit 10.1 |
|
January 10, 2014 |
|
|
|
|
|
|
|
|
|
|
10.17 |
|
Amendment, dated July 6, 2015, to Letter Agreement with Joseph Kennedy, dated December 13, 2011* |
|
Quarterly Report on Form 10-Q for the period ended June 30, 2015, File No. 0-21392, as Exhibit 10.1 |
|
August 6, 2015 |
|
|
|
|
|
|
|
|
|
10.18 |
|
Amendment, dated July 6, 2015, to Letter Agreement with Steven Ketchum, dated February 8, 2012* |
|
Quarterly Report on Form 10-Q for the period ended June 30, 2015, File No. 0-21392, as Exhibit 10.2 |
|
August 6, 2015 |
|
|
|
|
|
|
|
|
|
10.19 |
|
Amendment, dated July 6, 2015, to Letter Agreement with John Thero, dated December 23, 2011* |
|
Quarterly Report on Form 10-Q for the period ended June 30, 2015, File No. 0-21392, as Exhibit 10.3 |
|
August 6, 2015 |
|
|
|
|
|
|
|
|
|
10.20 |
|
2011 Long Term Incentive Award with Joseph Kennedy dated December 16, 2011* |
|
Form S-8, File No. 333-180180, as Exhibit 4.1 |
|
March 16, 2012 |
94
|
Exhibit |
|
|
|
Incorporated by Reference Herein |
||
|
Number |
|
Description |
|
Form |
|
Date |
|
|
|
|
|
|
|
|
|
10.21 |
|
2012 Long Term Incentive Award with Steven Ketchum dated March 1, 2012* |
|
Form S-8, File No. 333-180180, as Exhibit 4.2 |
|
March 16, 2012 |
|
|
|
|
|
|
|
|
|
10.22 |
|
Employment Agreement dated November 5, 2009 with John F. Thero* |
|
Registration Statement on Form F-1, File No. 333-163704, as Exhibit 4.104 |
|
December 14, 2009 |
|
|
|
|
|
|
|
|
|
10.23 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2007, File No. 0-21392, as Exhibit 4.67 |
|
May 19, 2008 |
|
|
|
|
|
|
|
|
|
|
10.24 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2008, File No. 0-21392, as Exhibit 4.90 |
|
October 22, 2009 |
|
|
|
|
|
|
|
|
|
|
10.25 |
|
Form of Purchase Agreement, dated June 1, 2007, between the Company and the Purchasers named therein |
|
Annual Report on Form 20-F for the year ended December 31, 2007, File No. 0-21392, as Exhibit 4.69 |
|
May 19, 2008 |
|
|
|
|
|
|
|
|
|
10.26 |
|
|
Report of Foreign Private Issuer filed on Form 6-K, File No. 0-21392, as Exhibit 99.5 |
|
December 17, 2007 |
|
|
|
|
|
|
|
|
|
|
10.27 |
|
|
Report of Foreign Private Issuer filed on Form 6-K, File No. 0-21392, as Exhibit 99.6 |
|
December 17, 2007 |
|
|
|
|
|
|
|
|
|
|
10.28 |
|
|
Report of Foreign Private Issuer filed on Form 6-K, File No. 0-21392, as Exhibit 99.7 |
|
December 17, 2007 |
|
|
|
|
|
|
|
|
|
|
10.29 |
|
|
Report of Foreign Private Issuer filed on Form 6-K, File No. 0-21392, as Exhibit 99.1 |
|
January 28, 2008 |
|
|
|
|
|
|
|
|
|
|
10.30 |
|
|
Report of Foreign Private Issuer filed on Form 6-K, File No. 0-21392, as Exhibit 99.1 |
|
February 1, 2008 |
|
|
|
|
|
|
|
|
|
|
10.31 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2007, File No. 0-21392, as Exhibit 4.79 |
|
May 19, 2008 |
|
|
|
|
|
|
|
|
|
|
10.32 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2008, File No. 0-21392, as Exhibit 4.80 |
|
October 22, 2009 |
|
|
|
|
|
|
|
|
|
|
10.33 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2007, File No. 0-21392, as Exhibit 4.81 |
|
May 19, 2008 |
|
|
|
|
|
|
|
|
|
|
10.34 |
|
|
Annual Report on Form 20-F/A for the year ended December 31, 2008, File No. 0-21392, as Exhibit 4.88 |
|
December 4, 2009 |
|
95
|
Exhibit |
|
|
|
Incorporated by Reference Herein |
||
|
Number |
|
Description |
|
Form |
|
Date |
|
|
|
|
|
|
|
|
|
10.35 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2008, File No. 0-21392, as Exhibit 4.94 |
|
October 22, 2009 |
|
|
|
|
|
|
|
|
|
|
10.36 |
|
|
Registration Statement on Form F-1, File No. 333-163704, as Exhibit 4.105 |
|
December 14, 2009 |
|
|
|
|
|
|
|
|
|
|
10.37 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2008, File No. 0-21392, as Exhibit 4.92 |
|
October 22, 2009 |
|
|
|
|
|
|
|
|
|
|
10.38 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2008, File No. 0-21392, as Exhibit 4.97 |
|
October 22, 2009 |
|
|
|
|
|
|
|
|
|
|
10.39 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2009, File No. 0-21392, as Exhibit 4.100 |
|
June 25, 2010 |
|
|
|
|
|
|
|
|
|
|
10.40 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2010, File No. 0-21392, as Exhibit 10.40 |
|
March 16, 2011 |
|
|
|
|
|
|
|
|
|
|
10.41 |
|
|
Quarterly Report on Form 10-Q for the period ended June 30, 2011, File No. 0-21392, as Exhibit 10.2 |
|
August 9, 2011 |
|
|
|
|
|
|
|
|
|
|
10.42 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended June 30, 2012, File No. 0-21392, as Exhibit 10.6 |
|
August 8, 2008 |
|
|
|
|
|
|
|
|
|
|
10.43 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended September 30, 2012, File No. 0-21392, as Exhibit 10.1 |
|
November 8, 2012 |
|
|
|
|
|
|
|
|
|
|
10.44 |
|
|
Quarterly Report on Form 10-Q for the period ended June 30, 2011, File No. 0-21392, as Exhibit 10.3 |
|
August 9, 2011 |
|
|
|
|
|
|
|
|
|
|
10.45 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended June 30, 2012, File No. 0-21392, as Exhibit 10.4 |
|
August 8, 2008 |
|
|
|
|
|
|
|
|
|
|
10.46 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended June 30, 2012, File No. 0-21392, as Exhibit 10.5 |
|
August 8, 2008 |
|
|
|
|
|
|
|
|
|
96
|
Exhibit |
|
|
|
Incorporated by Reference Herein |
||
|
Number |
|
Description |
|
Form |
|
Date |
|
|
|
|
|
|
|
|
|
10.47 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2012, File No. 0-21392, as Exhibit 10.71 |
|
February 28, 2012 |
|
|
|
|
|
|
|
|
|
|
10.48 |
|
|
Quarterly Report on Form 10-Q for the period ended September 30, 2011, File No. 0-21392, as Exhibit 10.2 |
|
November 8, 2011 |
|
|
|
|
|
|
|
|
|
|
10.49 |
|
|
Annual Report on Form 20-F for the year ended December 31, 2006, File No. 0-21392, as Exhibit 4.71 |
|
March 5, 2007 |
|
|
|
|
|
|
|
|
|
|
10.50 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2011, File No. 0-21392, as Exhibit 10.61 |
|
February 29, 2012 |
|
|
|
|
|
|
|
|
|
|
10.51 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended June 30, 2012, File No. 0-21392, as Exhibit 10.3 |
|
August 8, 2012 |
|
|
|
|
|
|
|
|
|
|
10.52 |
|
Lease Agreement dated May 8, 2013, by and between Amarin Pharma, Inc. and Bedminster 2 Funding, LLC. |
|
Quarterly Report on Form 10-Q for the period ended March 31, 2013, File No. 0-21392, as Exhibit 10.1 |
|
May 9, 2013 |
|
|
|
|
|
|
|
|
|
10.53 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2014, File No. 0-21392, as Exhibit 10.80 |
|
March 3, 2015 |
|
|
|
|
|
|
|
|
|
|
10.54 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2014, File No. 0-21392, as Exhibit 10.81 |
|
March 3, 2015 |
|
|
|
|
|
|
|
|
|
|
10.55 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2016, File No. 0-21392, as Exhibit 10.55 |
|
March 1, 2017 |
|
|
|
|
|
|
|
|
|
|
10.56 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2012, File No. 0-21392, as Exhibit 10.76 |
|
February 28, 2012 |
|
|
|
|
|
|
|
|
|
|
10.57 |
|
|
Quarterly Report on Form 10-Q for quarterly period ended March 31, 2014, File No. 0-21392, as Exhibit 10.1 |
|
May 9, 2014 |
|
|
|
|
|
|
|
|
|
|
10.58 |
|
|
Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2015, File No. 0-21392, as Exhibit 10.1 |
|
May 8, 2015 |
|
97
|
Exhibit |
|
|
|
Incorporated by Reference Herein |
||
|
Number |
|
Description |
|
Form |
|
Date |
|
|
|
|
|
|
|
|
|
10.59 |
|
|
Current Report on Form 8-K dated March 5, 2015, File No. 0-21392, File No. 0-21392, as Exhibit 10.1 |
|
March 11, 2015 |
|
|
|
|
|
|
|
|
|
|
10.60 |
|
|
Current Report on Form 8-K dated March 30, 2015, File No. 0-21392, as Exhibit 10.1 |
|
March 30, 2015 |
|
|
|
|
|
|
|
|
|
|
10.61 |
|
Letter Agreement, dated May 9, 2016, by and between Amarin Corporation plc and Michael Kalb* |
|
Current Report on Form 8-K dated June 30, 2016, File No. 0-21392, as Exhibit 10.1 |
|
June 30, 2016 |
|
|
|
|
|
|
|
|
|
10.62 |
|
|
Current Report on Form 8-K dated January 20, 2017, File No. 0-21392, as Exhibit 10.1. |
|
January 20, 2017 |
|
|
|
|
|
|
|
|
|
|
10.63 |
|
|
Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2017, File No. 0-21392, as Exhibit 4.1 |
|
August 2, 2017 |
|
|
|
|
|
|
|
|
|
|
10.64 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2017, File No. 0-21392, as Exhibit 10.64 |
|
February 27, 2018 |
|
|
|
|
|
|
|
|
|
|
10.65 |
|
|
Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2017, File No. 0-21392, as Exhibit 10.1 |
|
August 2, 2017 |
|
|
|
|
|
|
|
|
|
|
10.66 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2017, File No. 0-21392, as Exhibit 10.66 |
|
February 27, 2018 |
|
|
|
|
|
|
|
|
|
|
10.67 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2017, File No. 0-21392, as Exhibit 10.67 |
|
February 27, 2018 |
|
|
|
|
|
|
|
|
|
|
10.68 |
|
|
Annual Report on Form 10-K for the year ended December 31, 2017, File No. 0-21392, as Exhibit 10.68 |
|
February 27, 2018 |
|
|
|
|
|
|
|
|
|
|
10.69 |
|
Lease Agreement, dated February 5, 2019, by and between 440 Route 22 LLC and Amarin Pharma, Inc. |
|
Filed herewith |
|
|
|
|
|
|
|
|
|
|
|
14.1 |
|
|
Registration Statement on Form F-3, File No. 333-170505, as Exhibit 99.1 |
|
November 10, 2010 |
|
98
|
Exhibit |
|
|
|
Incorporated by Reference Herein |
||
|
Number |
|
Description |
|
Form |
|
Date |
|
|
|
|
|
|
|
|
|
21.1 |
|
|
Filed herewith |
|
|
|
|
|
|
|
|
|
|
|
|
23.1 |
|
|
Filed herewith |
|
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
|
Filed herewith |
|
|
|
|
|
|
|
|
|
|
|
|
31.2 |
|
|
Filed herewith |
|
|
|
|
|
|
|
|
|
|
|
|
32.1 |
|
|
Filed herewith |
|
|
|
|
|
|
|
|
|
|
|
|
101 |
|
INS XBRL Instance Document |
|
|
|
|
|
|
|
|
|
|
|
|
|
101 |
|
SCH XBRL Taxonomy Extension Schema Document |
|
|
|
|
|
|
|
|
|
|
|
|
|
101 |
|
CAL XBRL Taxonomy Calculation Linkbase Document |
|
|
|
|
|
|
|
|
|
|
|
|
|
101 |
|
DEF XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
|
|
|
|
|
|
|
|
|
|
101 |
|
LAB XBRL Taxonomy Label Linkbase Document |
|
|
|
|
|
|
|
|
|
|
|
|
|
101 |
|
PRE XBRL Taxonomy Presentation Linkbase Document |
|
|
|
|
|
†† |
Confidential treatment has been granted with respect to portions of this exhibit pursuant to an application requesting confidential treatment under Rule 24b-2 of the Securities Exchange Act of 1934. A complete copy of this exhibit, including the redacted terms, has been separately filed with the Securities and Exchange Commission. |
|
* |
Management contract or compensatory plan or arrangement. |
Not applicable.
99
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
|
|
|
AMARIN CORPORATION PLC |
||
|
|
|
|
|
By: |
|
|
|
|
/s/ John F. Thero
|
|
|
|
John F. Thero |
|
|
|
President and Chief Executive Officer (Principal Executive Officer) |
|
Date: February 27, 2019
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the date indicated.
|
|
|
|
|
Signature
|
Title
|
Date
|
|
|
|
|
|
/s/ John F. Thero
John F. Thero |
Director, President and Chief Executive Officer (Principal Executive Officer) |
February 27, 2019 |
|
|
|
|
|
/s/ Michael W. Kalb
Michael W. Kalb |
Senior Vice President and Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) |
February 27, 2019 |
|
|
|
|
|
/s/ Lars Ekman, M.D., Ph.D.
Lars Ekman, M.D., Ph.D. |
Director |
February 27, 2019 |
|
|
|
|
|
/s/ Patrick O’Sullivan
Patrick O’Sullivan |
Director |
February 27, 2019 |
|
|
|
|
|
/s/ Kristine Peterson
Kristine Peterson |
Director |
February 27, 2019 |
|
|
|
|
|
/s/ David Stack
David Stack |
Director |
February 27, 2019 |
|
|
|
|
|
/s/ Jan van Heek
Jan van Heek |
Director |
February 27, 2019 |
|
|
|
|
|
/s/ Joseph Zakrzewski
Joseph Zakrzewski |
Director |
February 27, 2019 |
100
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
|
Page |
|
|
F-2 |
|
|
Financial Statements: |
|
|
|
Consolidated Balance Sheets as of December 31, 2018 and 2017 |
|
F-3 |
|
Consolidated Statements of Operations for the years ended December 31, 2018, 2017 and 2016 |
|
F-4 |
|
|
F-5 |
|
|
Consolidated Statements of Cash Flows for the years ended December 31, 2018, 2017 and 2016 |
|
F-6 |
|
|
F-7 |
|
|
|
|
|
|
Financial Statement Schedules: |
|
|
Financial statement schedules have been omitted for the reason that the required information is presented in the consolidated financial statements or notes thereto, the amounts involved are not significant or the schedules are not applicable.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors of Amarin Corporation plc
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Amarin Corporation plc (the Company) as of December 31, 2018 and 2017, the related consolidated statements of operations, stockholders’ deficit and cash flows for each of the three years in the period ended December 31, 2018 and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2018 and 2017 and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2018, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2018, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 27, 2019 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2014.
Iselin, New Jersey
February 27, 2019
F-2
(in thousands, except share amounts)
|
|
|
As of December 31, |
|
|||||
|
|
|
2018 |
|
|
2017 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
|
Current Assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
249,227 |
|
|
$ |
73,637 |
|
|
Restricted cash |
|
|
1,500 |
|
|
|
600 |
|
|
Accounts receivable, net |
|
|
66,523 |
|
|
|
45,318 |
|
|
Inventory |
|
|
57,802 |
|
|
|
30,260 |
|
|
Prepaid and other current assets |
|
|
2,945 |
|
|
|
3,455 |
|
|
Total current assets |
|
|
377,997 |
|
|
|
153,270 |
|
|
Property, plant and equipment, net |
|
|
63 |
|
|
|
28 |
|
|
Other long-term assets |
|
|
174 |
|
|
|
174 |
|
|
Intangible asset, net |
|
|
7,480 |
|
|
|
8,126 |
|
|
TOTAL ASSETS |
|
$ |
385,714 |
|
|
$ |
161,598 |
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) |
|
|
|
|
|
|
|
|
|
Current Liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
37,632 |
|
|
$ |
25,155 |
|
|
Accrued expenses and other current liabilities |
|
|
84,171 |
|
|
|
58,902 |
|
|
Current portion of exchangeable senior notes, net of discount |
|
|
— |
|
|
|
481 |
|
|
Current portion of long-term debt from royalty-bearing instrument |
|
|
34,240 |
|
|
|
22,348 |
|
|
Deferred revenue, current |
|
|
1,220 |
|
|
|
1,644 |
|
|
Total current liabilities |
|
|
157,263 |
|
|
|
108,530 |
|
|
Long-Term Liabilities: |
|
|
|
|
|
|
|
|
|
Exchangeable senior notes, net of discount |
|
|
— |
|
|
|
28,992 |
|
|
Long-term debt from royalty-bearing instrument |
|
|
46,108 |
|
|
|
70,834 |
|
|
Deferred revenue, long-term |
|
|
19,490 |
|
|
|
17,192 |
|
|
Other long-term liabilities |
|
|
10,523 |
|
|
|
1,150 |
|
|
Total liabilities |
|
|
233,384 |
|
|
|
226,698 |
|
|
Commitments and contingencies (Note 8) |
|
|
|
|
|
|
|
|
|
Stockholders’ Equity (Deficit): |
|
|
|
|
|
|
|
|
|
Series A Convertible Preferred Stock, £0.05 par, unlimited authorized; 289,317,460 shares issued and outstanding as of December 31, 2018 (equivalent to 28,931,746 ordinary shares upon future consolidation and redesignation at a 10:1 ratio); 328,184,640 shares issued and outstanding as of December 31, 2017 (equivalent to 32,818,464 ordinary shares upon future consolidation and redesignation at a 10:1 ratio) |
|
|
21,850 |
|
|
|
24,364 |
|
|
Common stock, £0.50 par, unlimited authorized; 329,110,863 issued, 325,850,013 outstanding as of December 31, 2018; 272,719,044 issued, 271,022,011 outstanding as of December 31, 2017 |
|
|
246,663 |
|
|
|
208,768 |
|
|
Additional paid-in capital |
|
|
1,282,762 |
|
|
|
977,866 |
|
|
Treasury stock; 3,260,850 shares as of December 31, 2018; 1,697,033 shares as of December 31, 2017 |
|
|
(10,413 |
) |
|
|
(4,229 |
) |
|
Accumulated deficit |
|
|
(1,388,532 |
) |
|
|
(1,271,869 |
) |
|
Total stockholders’ equity (deficit) |
|
|
152,330 |
|
|
|
(65,100 |
) |
|
TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) |
|
$ |
385,714 |
|
|
$ |
161,598 |
|
See the notes to the consolidated financial statements.
F-3
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
|
|
Year Ended December 31, |
|
|||||||||
|
|
2018 |
|
|
2017 |
|
|
2016 |
|
|||
|
Product revenue, net |
$ |
228,371 |
|
|
$ |
179,825 |
|
|
$ |
128,966 |
|
|
Licensing revenue |
|
843 |
|
|
|
1,279 |
|
|
|
1,118 |
|
|
Total revenue, net |
|
229,214 |
|
|
|
181,104 |
|
|
|
130,084 |
|
|
Less: Cost of goods sold |
|
54,543 |
|
|
|
44,952 |
|
|
|
34,363 |
|
|
Gross margin |
|
174,671 |
|
|
|
136,152 |
|
|
|
95,721 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
Selling, general and administrative |
|
226,996 |
|
|
|
134,549 |
|
|
|
111,372 |
|
|
Research and development |
|
55,900 |
|
|
|
47,158 |
|
|
|
49,975 |
|
|
Total operating expenses |
|
282,896 |
|
|
|
181,707 |
|
|
|
161,347 |
|
|
Operating loss |
|
(108,225 |
) |
|
|
(45,555 |
) |
|
|
(65,626 |
) |
|
Gain on change in fair value of derivative liabilities |
|
— |
|
|
|
— |
|
|
|
8,170 |
|
|
Interest expense |
|
(8,872 |
) |
|
|
(9,766 |
) |
|
|
(18,677 |
) |
|
Interest income |
|
1,074 |
|
|
|
429 |
|
|
|
234 |
|
|
Other (expense) income, net |
|
(326 |
) |
|
|
74 |
|
|
|
(482 |
) |
|
Loss from operations before taxes |
|
(116,349 |
) |
|
|
(54,818 |
) |
|
|
(76,381 |
) |
|
Provision for income taxes |
|
(96 |
) |
|
|
(13,047 |
) |
|
|
(9,969 |
) |
|
Net loss |
|
(116,445 |
) |
|
|
(67,865 |
) |
|
|
(86,350 |
) |
|
Loss per share: |
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
$ |
(0.39 |
) |
|
$ |
(0.25 |
) |
|
$ |
(0.41 |
) |
|
Diluted |
$ |
(0.39 |
) |
|
$ |
(0.25 |
) |
|
$ |
(0.41 |
) |
|
Weighted average shares outstanding: |
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
297,237 |
|
|
|
270,652 |
|
|
|
211,874 |
|
|
Diluted |
|
297,237 |
|
|
|
270,652 |
|
|
|
211,874 |
|
See the notes to the consolidated financial statements.
F-4
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
(in thousands, except share amounts)
|
|
|
Preferred Shares |
|
|
Common Shares |
|
|
Treasury Shares |
|
|
Preferred Stock |
|
|
Common Stock |
|
|
Additional Paid-in Capital |
|
|
Treasury Stock |
|
|
Accumulated Deficit |
|
|
Total |
|
|||||||||
|
December 31, 2015 |
|
|
328,184,640 |
|
|
|
183,577,765 |
|
|
|
(174,502 |
) |
|
$ |
24,364 |
|
|
$ |
149,978 |
|
|
$ |
816,171 |
|
|
$ |
(411 |
) |
|
$ |
(1,119,293 |
) |
|
$ |
(129,191 |
) |
|
Cumulative-effect adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,639 |
|
|
|
1,639 |
|
|
January 1, 2016 |
|
|
328,184,640 |
|
|
|
183,577,765 |
|
|
|
(174,502 |
) |
|
$ |
24,364 |
|
|
$ |
149,978 |
|
|
$ |
816,171 |
|
|
$ |
(411 |
) |
|
$ |
(1,117,654 |
) |
|
$ |
(127,552 |
) |
|
Issuance of common stock, net of transaction costs |
|
|
— |
|
|
|
24,265,000 |
|
|
|
— |
|
|
|
— |
|
|
|
15,712 |
|
|
|
48,901 |
|
|
|
— |
|
|
|
— |
|
|
|
64,613 |
|
|
Exchange of exchangeable senior notes, net of transaction costs |
|
|
— |
|
|
|
60,311,188 |
|
|
|
— |
|
|
|
— |
|
|
|
40,062 |
|
|
|
87,374 |
|
|
|
— |
|
|
|
— |
|
|
|
127,436 |
|
|
Exercise of stock options |
|
|
— |
|
|
|
177,146 |
|
|
|
— |
|
|
|
— |
|
|
|
119 |
|
|
|
168 |
|
|
|
— |
|
|
|
— |
|
|
|
287 |
|
|
Vesting of restricted stock units |
|
|
— |
|
|
|
1,852,102 |
|
|
|
(645,003 |
) |
|
|
— |
|
|
|
1,295 |
|
|
|
(1,302 |
) |
|
|
(1,087 |
) |
|
|
— |
|
|
|
(1,094 |
) |
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
13,602 |
|
|
|
— |
|
|
|
— |
|
|
|
13,602 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(86,350 |
) |
|
|
(86,350 |
) |
|
December 31, 2016 |
|
|
328,184,640 |
|
|
|
270,183,201 |
|
|
|
(819,505 |
) |
|
$ |
24,364 |
|
|
$ |
207,166 |
|
|
$ |
964,914 |
|
|
$ |
(1,498 |
) |
|
$ |
(1,204,004 |
) |
|
$ |
(9,058 |
) |
|
Exercise of stock options |
|
|
— |
|
|
|
356,656 |
|
|
|
— |
|
|
|
— |
|
|
|
229 |
|
|
|
409 |
|
|
|
— |
|
|
|
— |
|
|
|
638 |
|
|
Vesting of restricted stock units |
|
|
— |
|
|
|
2,179,187 |
|
|
|
(877,528 |
) |
|
|
— |
|
|
|
1,373 |
|
|
|
(1,409 |
) |
|
|
(2,731 |
) |
|
|
— |
|
|
|
(2,767 |
) |
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
13,952 |
|
|
|
— |
|
|
|
— |
|
|
|
13,952 |
|
|
Loss for the period |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(67,865 |
) |
|
|
(67,865 |
) |
|
December 31, 2017 |
|
|
328,184,640 |
|
|
|
272,719,044 |
|
|
|
(1,697,033 |
) |
|
$ |
24,364 |
|
|
$ |
208,768 |
|
|
$ |
977,866 |
|
|
$ |
(4,229 |
) |
|
$ |
(1,271,869 |
) |
|
$ |
(65,100 |
) |
|
Cumulative-effect adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(218 |
) |
|
|
(218 |
) |
|
January 1, 2018 |
|
|
328,184,640 |
|
|
|
272,719,044 |
|
|
|
(1,697,033 |
) |
|
$ |
24,364 |
|
|
$ |
208,768 |
|
|
$ |
977,866 |
|
|
$ |
(4,229 |
) |
|
$ |
(1,272,087 |
) |
|
$ |
(65,318 |
) |
|
Issuance of common stock, net of transaction costs |
|
|
— |
|
|
|
31,727,550 |
|
|
|
— |
|
|
|
— |
|
|
|
21,744 |
|
|
|
243,096 |
|
|
|
— |
|
|
|
— |
|
|
|
264,840 |
|
|
Issuance of common stock under employee stock purchase plan |
|
|
— |
|
|
|
312,257 |
|
|
|
— |
|
|
|
— |
|
|
|
203 |
|
|
|
840 |
|
|
|
— |
|
|
|
— |
|
|
|
1,043 |
|
|
Exchange of exchangeable senior notes, net of transaction costs |
|
|
— |
|
|
|
7,716,046 |
|
|
|
— |
|
|
|
— |
|
|
|
5,011 |
|
|
|
24,358 |
|
|
|
— |
|
|
|
— |
|
|
|
29,369 |
|
|
Conversion of Series A Convertible Preferred Stock, net |
|
|
(38,867,180 |
) |
|
|
3,886,718 |
|
|
|
— |
|
|
|
(2,514 |
) |
|
|
2,514 |
|
|
|
(39 |
) |
|
|
— |
|
|
|
— |
|
|
|
(39 |
) |
|
Exercise of stock options |
|
|
— |
|
|
|
8,138,305 |
|
|
|
— |
|
|
|
— |
|
|
|
5,309 |
|
|
|
21,093 |
|
|
|
— |
|
|
|
— |
|
|
|
26,402 |
|
|
Vesting of restricted stock units |
|
|
— |
|
|
|
4,610,943 |
|
|
|
(1,563,817 |
) |
|
|
— |
|
|
|
3,114 |
|
|
|
(3,114 |
) |
|
|
(6,184 |
) |
|
|
— |
|
|
|
(6,184 |
) |
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
18,662 |
|
|
|
— |
|
|
|
— |
|
|
|
18,662 |
|
|
Loss for the period |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(116,445 |
) |
|
|
(116,445 |
) |
|
December 31, 2018 |
|
|
289,317,460 |
|
|
|
329,110,863 |
|
|
|
(3,260,850 |
) |
|
$ |
21,850 |
|
|
$ |
246,663 |
|
|
$ |
1,282,762 |
|
|
$ |
(10,413 |
) |
|
$ |
(1,388,532 |
) |
|
$ |
152,330 |
|
See the notes to the consolidated financial statements.
F-5
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2018 |
|
|
2017 |
|
|
2016 |
|
|||
|
CASH FLOWS FROM OPERATING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(116,445 |
) |
|
$ |
(67,865 |
) |
|
$ |
(86,350 |
) |
|
Adjustments to reconcile loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
23 |
|
|
|
62 |
|
|
|
138 |
|
|
Loss on sale of fixed assets |
|
|
— |
|
|
|
— |
|
|
|
48 |
|
|
Allowance for doubtful accounts |
|
|
— |
|
|
|
— |
|
|
|
12 |
|
|
Stock-based compensation |
|
|
18,806 |
|
|
|
13,960 |
|
|
|
13,613 |
|
|
Amortization of debt discount and debt issuance costs |
|
|
2,183 |
|
|
|
2,332 |
|
|
|
7,783 |
|
|
Amortization of intangible asset |
|
|
646 |
|
|
|
646 |
|
|
|
645 |
|
|
Gain on change in fair value of derivative liabilities |
|
|
— |
|
|
|
— |
|
|
|
(8,170 |
) |
|
Deferred income taxes |
|
|
— |
|
|
|
11,082 |
|
|
|
8,798 |
|
|
Changes in assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable, net |
|
|
(21,205 |
) |
|
|
(25,333 |
) |
|
|
(6,171 |
) |
|
Inventory |
|
|
(27,542 |
) |
|
|
(9,753 |
) |
|
|
(1,522 |
) |
|
Prepaid and other current assets |
|
|
510 |
|
|
|
6,028 |
|
|
|
(3,831 |
) |
|
Other long-term assets |
|
|
— |
|
|
|
567 |
|
|
|
(567 |
) |
|
Accrued interest payable |
|
|
(310 |
) |
|
|
(6,491 |
) |
|
|
(6,205 |
) |
|
Deferred revenue |
|
|
1,656 |
|
|
|
1,221 |
|
|
|
884 |
|
|
Accounts payable and other current liabilities |
|
|
37,602 |
|
|
|
40,267 |
|
|
|
8,705 |
|
|
Other long-term liabilities |
|
|
9,373 |
|
|
|
440 |
|
|
|
375 |
|
|
Net cash used in operating activities |
|
|
(94,703 |
) |
|
|
(32,837 |
) |
|
|
(71,815 |
) |
|
CASH FLOWS FROM INVESTING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of equipment |
|
|
(58 |
) |
|
|
(12 |
) |
|
|
(21 |
) |
|
Net cash used in investing activities |
|
|
(58 |
) |
|
|
(12 |
) |
|
|
(21 |
) |
|
CASH FLOWS FROM FINANCING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of exchangeable debt |
|
|
— |
|
|
|
30,000 |
|
|
|
— |
|
|
Proceeds from issuance of common stock, net of transaction costs |
|
|
264,840 |
|
|
|
— |
|
|
|
64,613 |
|
|
Proceeds from issuance of common stock under employee stock purchase plan |
|
|
1,043 |
|
|
|
— |
|
|
|
— |
|
|
Proceeds from exercise of stock options, net of transaction costs |
|
|
26,402 |
|
|
|
638 |
|
|
|
287 |
|
|
Payment of debt issuance costs |
|
|
— |
|
|
|
(1,207 |
) |
|
|
— |
|
|
Payment of transaction costs for conversion of preferred stock |
|
|
(39 |
) |
|
|
— |
|
|
|
— |
|
|
Repurchase of exchangeable senior notes, including transaction costs |
|
|
— |
|
|
|
(15,107 |
) |
|
|
— |
|
|
Payment on long-term debt from royalty-bearing instrument |
|
|
(14,690 |
) |
|
|
(3,322 |
) |
|
|
— |
|
|
Transaction costs related to exchange of exchangeable senior notes |
|
|
(121 |
) |
|
|
— |
|
|
|
(680 |
) |
|
Taxes paid related to stock-based awards |
|
|
(6,184 |
) |
|
|
(2,767 |
) |
|
|
(1,094 |
) |
|
Net cash provided by financing activities |
|
|
271,251 |
|
|
|
8,235 |
|
|
|
63,126 |
|
|
NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS AND RESTRICTED CASH |
|
|
176,490 |
|
|
|
(24,614 |
) |
|
|
(8,710 |
) |
|
CASH AND CASH EQUIVALENTS AND RESTRICTED CASH, BEGINNING OF PERIOD |
|
|
74,237 |
|
|
|
98,851 |
|
|
|
107,561 |
|
|
CASH AND CASH EQUIVALENTS AND RESTRICTED CASH, END OF PERIOD |
|
$ |
250,727 |
|
|
$ |
74,237 |
|
|
$ |
98,851 |
|
|
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid during the year for: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest |
|
$ |
21,527 |
|
|
$ |
17,241 |
|
|
$ |
17,083 |
|
|
Income taxes |
|
$ |
850 |
|
|
$ |
1,753 |
|
|
$ |
1,457 |
|
|
Supplemental disclosure of non-cash transactions: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Exchange of exchangeable senior notes into common stock |
|
$ |
29,490 |
|
|
$ |
— |
|
|
$ |
128,115 |
|
|
Conversion of Series A Convertible Preferred Stock into common stock |
|
$ |
2,514 |
|
|
$ |
— |
|
|
$ |
— |
|
See the notes to the consolidated financial statements.
AMARIN CORPORATION PLC
F-6
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(1) Nature of Business and Basis of Presentation
Nature of Business
Amarin Corporation plc (“Amarin” or the “Company”) is a pharmaceutical company with expertise in omega-3 fatty acids and lipid science focused on the commercialization and development of therapeutics to improve cardiovascular health and reduce cardiovascular risk.
The Company’s lead product, Vascepa® (icosapent ethyl) capsules, is approved by the U.S. Food and Drug Administration, or FDA, for use as an adjunct to diet to reduce triglyceride levels in adult patients with severe (TG ≥500 mg/dL) hypertriglyceridemia. Vascepa is available in the United States by prescription only. In January 2013, the Company began selling and marketing 1-gram size Vascepa capsules in the United States, and in October 2016, introduced a smaller 0.5-gram capsule size. In August 2015, in addition to marketing Vascepa for severe hypertriglyceridemia, the Company commenced marketing Vascepa for use in adult patients with mixed dyslipidemia, as an adjunct to diet and an add-on to statin therapy in patients who despite statin therapy have high triglycerides (TGs ≥200 mg/dL and ≤500 mg/dL), which the Company also refers to as persistently high triglycerides. This expanded promotion of Vascepa commenced pursuant to a federal court order and is continuing pursuant to an agreement among the Company, the FDA and the U.S. government.
The Company also developed Vascepa for FDA approval of potential additional indications for use. In particular, the Company conducted a cardiovascular outcomes study of Vascepa, titled REDUCE-IT™ (Reduction of Cardiovascular Events with EPA—Intervention Trial). The REDUCE-IT study, which commenced in 2011 and completed patient enrollment and randomization of 8,179 individual patients in 2016, was designed to evaluate the efficacy of Vascepa in reducing major cardiovascular events in a high-risk patient population on statin therapy. The REDUCE-IT study topline results were made public in September 2018, and the primary results of the REDUCE-IT study were presented at the 2018 Scientific Sessions of the American Heart Association (AHA) in November 2018 with such results concurrently published in The New England Journal of Medicine.
The Company sells Vascepa principally to a limited number of major wholesalers, as well as selected regional wholesalers and specialty pharmacy providers, or collectively, its Distributors or its customers, that in turn resell Vascepa to retail pharmacies for subsequent resale to patients and healthcare providers. The Company markets Vascepa in the United States through its direct sales force, which prior to the REDUCE-IT results topline announcement in September 2018 consisted of approximately 170 sales professionals, including sales representatives and their managers, and increased to approximately 440 sales professionals, including approximately 400 sales representatives, to begin 2019. The Company plans to submit an sNDA to the FDA seeking revised labeling for Vascepa based on results of the REDUCE-IT study and, upon such expanded labeling, subject to FDA approval of such label, to further expand its promotion of Vascepa in the United States. Since its commencement in May 2014 until its scheduled expiration on December 31, 2018, Vascepa was co-promoted in the United States under an agreement with Kowa Pharmaceuticals America, Inc. Outside of the United States, the Company has entered into agreements with third party companies in select geographies for purposes of pursuing regulatory approval and commercialization of Vascepa. The Company operates in one business segment.
Basis of Presentation
The consolidated financial statements included herein have been prepared by the Company in accordance with accounting principles generally accepted in the United States of America (the “U.S.” or the “United States”) and pursuant to the rules and regulations of the Securities and Exchange Commission, or the SEC.
The consolidated financial statements reflect all adjustments of a normal and recurring nature that, in the opinion of management, are necessary to present fairly the Company’s financial position, results of operations and cash flows for the periods indicated. The preparation of the Company’s consolidated financial statements in conformity with U.S. Generally Accepted Accounting Principles (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. The results of operations for the years ended December 31, 2018, 2017 and 2016 are not necessarily indicative of the results for any future period. Certain numbers presented throughout this document may not add precisely to the totals provided due to rounding. Absolute and percentage changes are calculated using the underlying amounts in thousands. Certain prior year balances related to beginning and ending cash and cash equivalents and restricted cash in the consolidated statements of cash flows have been conformed to the current year presentation.
The accompanying consolidated financial statements of the Company and subsidiaries have been prepared on a basis which assumes that the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business.
F-7
At December 31, 2018, the Company had current assets of $378.0 million, including cash and cash equivalents of $249.2 million, accounts receivable, net, of $66.5 million and inventory, net, of $57.8 million. The Company’s consolidated balance sheet at December 31, 2018 also includes long-term debt from a royalty-bearing instrument which is anticipated to be repaid quarterly calculated as a percentage of Vascepa net revenues until fully satisfied. As of December 31, 2018, the Company had no other debt outstanding. In January 2017, the Company issued $30.0 million in aggregate principal amount of January 2017 3.5% exchangeable senior notes due 2047, or the 2017 Notes. The terms of the 2017 Notes contained a provision that allowed the Company to elect at its option to cause all or any portion of the 2017 Notes to be mandatorily exchanged into American Depositary Shares (“ADSs”) upon satisfaction of specified equity conditions. In October 2018, the Company exercised that option and mandatorily exchanged the entirety of the 2017 Notes into ADSs resulting in elimination of the debt, such that no amount of 2017 Notes remained as of December 31, 2018. The 2017 Notes were such that they could have been redeemed by the Company for cash on or after January 19, 2021, could have been put back to the Company by the holders on January 19, 2022 for cash equal to 100% of the principal amount plus any accrued and unpaid interest, and could have been exchanged into ADSs at the option of holders at any time after issuance and prior to maturity. Accordingly, the 2017 Notes did not represent a short-term claim on the liquid assets of the Company as of December 31, 2017.
The Company believes its cash and cash equivalents will be sufficient to fund its projected operations for at least twelve months and through the likely Prescription Drug User Fee Act (PDUFA) date for approval of a supplemental new drug application (sNDA) by the FDA based on REDUCE-IT study results. Depending on the level of cash generated from operations, and depending in part on the timing and results of the FDA review of the sNDA and rate of prescription growth for Vascepa, additional capital may be required support planned expansion of Vascepa promotion and potential Vascepa promotion beyond which the Company is currently executing. If additional capital is required and the Company is unable to obtain additional capital, the Company may be forced to delay, limit or eliminate certain promotional activities. The Company anticipates that annual net cash outflows in future periods will be variable.
(2) Significant Accounting Policies
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates
Accounting estimates are based on historical experience and other factors that are considered reasonable under the circumstances. Estimates are used in determining such items as provisions for sales returns, rebates and incentives, chargebacks, and other sales allowances; depreciable/amortizable lives; asset impairments; valuation allowance on deferred taxes; probabilities of achievement of performance conditions for certain equity awards; amounts recorded for licensing revenue; contingencies and accruals; and valuations of derivative and long-term debt instruments. Because of the uncertainties inherent in such estimates, actual results may differ from these estimates. Management periodically evaluates estimates used in the preparation of the consolidated financial statements for continued reasonableness.
Use of Forecasted Financial Information in Accounting Estimates
The use of forecasted financial information is inherent in many of the Company’s accounting estimates including, but not limited to, determining the estimated fair values of derivatives, debt instruments and intangible assets, evaluating the need for valuation allowances for deferred tax assets, and assessing the Company’s ability to continue as a going concern. Such forecasted financial information is comprised of numerous assumptions regarding the Company’s future revenues, cash flows, and operational results. Management believes that its financial forecasts are reasonable and appropriate based upon current facts and circumstances. Because of the inherent nature of forecasts, however, actual results may differ from these forecasts. Management regularly reviews the information related to these forecasts and adjusts the carrying amounts of the applicable assets prospectively, if and when actual results differ from previous estimates.
Revenue Recognition
Effective January 1, 2018, the Company adopted Accounting Standards Codification, or ASC, Topic 606, Revenue from Contracts with Customers, using the modified retrospective transition method. Under this method, the Company revised its opening retained earnings balance with a cumulative-effect adjustment of $0.2 million related solely to licensing revenues as of January 1, 2018. This standard applies to all contracts with customers except for contracts that are within the scope of other standards, such as leases, insurance, and financial instruments. Under Topic 606, an entity recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that an entity determines are within the scope of Topic 606, the entity
F-8
performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the entity will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer. At contract inception, once the contract is determined to be within the scope of Topic 606, the Company assesses the goods or services promised within each contract and determines those that are performance obligations and assesses whether each promised good or service is distinct. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied. For a complete discussion of accounting for net product revenue and licensing revenue, see Note 15—Revenue Recognition.
Distribution Costs
The Company records distribution costs related to shipping product to its customers, primarily through the use of common carriers or external distribution services, in cost of goods sold.
Cash and Cash Equivalents and Restricted Cash
Cash and cash equivalents consist of cash, deposits with banks and short-term highly liquid money market instruments with remaining maturities at the date of purchase of 90 days or less. Restricted cash represents cash and cash equivalents pledged to guarantee repayment of certain expenses which may be incurred for business travel under corporate credit cards held by employees.
Accounts Receivable, net
Accounts receivable, net, comprised of trade receivables, are generally due within 30 days and are stated at amounts due from customers. The Company recognizes an allowance for losses on accounts receivable in an amount equal to the estimated probable losses net of any recoveries. The allowance is based primarily on assessment of specific identifiable customer accounts considered at risk or uncollectible, as well as an analysis of current receivables aging and expected future write-offs. The expense associated with the allowance for doubtful accounts is recognized as selling, general, and administrative expense. The Company has not historically experienced any significant credit losses.
The following table summarizes the impact of accounts receivable reserves on the gross trade accounts receivable balances as of December 31, 2018 and 2017:
|
In thousands |
|
December 31, 2018 |
|
|
December 31, 2017 |
|
||
|
Gross trade accounts receivable |
|
$ |
86,133 |
|
|
$ |
57,802 |
|
|
Trade allowances |
|
|
(19,495 |
) |
|
|
(12,035 |
) |
|
Chargebacks |
|
|
(115 |
) |
|
|
(449 |
) |
|
Accounts receivable, net |
|
$ |
66,523 |
|
|
$ |
45,318 |
|
Inventory
The Company states inventories at the lower of cost or net realizable value. Cost is determined based on actual cost using the average cost method. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. An allowance is established when management determines that certain inventories may not be saleable. If inventory cost exceeds expected net realizable value due to obsolescence, damage or quantities in excess of expected demand, changes in price levels or other causes, the Company will reduce the carrying value of such inventory to net realizable value and recognize the difference as a component of cost of goods sold in the period in which it occurs. The Company capitalizes inventory purchases of saleable product from approved suppliers while inventory purchases from suppliers prior to regulatory approval are included as a component of research and development expense. The Company expenses inventory identified for use as marketing samples when they are packaged. The average cost reflects the actual purchase price of Vascepa active pharmaceutical ingredient, or API.
F-9
The Company provides for depreciation and amortization using the straight-line method by charges to operations in amounts that depreciate the cost of the fixed asset over its estimated useful life. The estimated useful lives, by asset classification, are as follows:
|
|
Useful Lives |
|
|
Computer equipment and software |
|
3 - 5 years |
|
Furniture and fixtures |
|
5 years |
|
Leasehold improvements |
|
Lesser of useful life or lease term |
Upon retirement or sale of assets, the cost of the assets disposed and the related accumulated depreciation are removed from the consolidated balance sheet and any resulting gain or loss is credited or expensed to operations. Repairs and maintenance costs are expensed as incurred.
Long-Lived Asset Impairment
The Company reviews its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Recoverability of these assets is determined by comparing the forecasted undiscounted net cash flows of the operation to which the assets relate to their carrying amount. If impairment is indicated, the assets are written down to fair value. Fair value is determined based on discounted forecasted cash flows or appraised values, depending on the nature of the assets.
Intangible Asset, net
Intangible asset, net consists of a milestone payment paid to the former shareholders of Laxdale Limited related to the 2004 acquisition of the rights to Vascepa, which is the result of Vascepa receiving marketing approval for the first indication and is amortized over its estimated useful life on a straight-line basis. See Note 8—Commitments and Contingencies for further information regarding other obligations related to the acquisition of Laxdale Limited.
Costs for Patent Litigation and Legal Proceedings
Costs for patent litigation or other legal proceedings are expensed as incurred and included in selling, general and administrative expenses.
Research and Development Costs
The Company charges research and development costs to operations as incurred. Research and development expenses are comprised of costs incurred by the Company in performing research and development activities, including: salary and benefits; stock-based compensation expense; laboratory supplies and other direct expenses; contractual services, including clinical trial and pharmaceutical development costs; commercial supply investment in its drug candidates; and infrastructure costs, including facilities costs and depreciation expense. In addition, research and development costs include the costs of product supply received from suppliers when such receipt by the Company is prior to regulatory approval of the supplier, as well as license fees related to the Company’s strategic collaboration with Mochida Pharmaceutical Co., Ltd.
Selling, General and Administrative Costs
The Company charges selling, general and administrative costs to operations as incurred. Selling, general and administrative costs include salaries and benefits, stock-based compensation expense, and costs of programs and infrastructure necessary for the general conduct of the Company’s business, including those incurred as a result of the commercialization of Vascepa in the United States as well as co-promotion fees to Kowa Pharmaceuticals America, Inc. which in 2018 included costs for accrual of tail co-promotion fees.
Income Taxes
On December 22, 2017, the U.S. enacted the Tax Cuts and Jobs Act (the “Act”) which instituted fundamental changes to the taxation of multinational corporations. The Act includes changes to the taxation of foreign earnings by implementing a dividend exemption system, expansion of the current anti-deferral rules, a minimum tax on low-taxed foreign earnings and new measures to deter base erosion. The Act also includes a permanent reduction in the corporate tax rate to 21%, repeal of the corporate alternative minimum tax, expensing of capital investment, and limitation of the deduction of interest expense. Furthermore, as part of the transition to the new tax system, a one-time transition tax is imposed on a U.S. shareholder's historical undistributed earnings of foreign affiliates. The Company applied the guidance in Staff Accounting Bulletin 118 when accounting for the enactment-date effects of the Act in the prior
F-10
year. As of December 31, 2017, the Company had recorded provisional amounts to account for the impact of tax effects of the Act related to the change in corporate tax rate from 34% to 21% and the changes to executive compensation deductibility. As of December 31, 2018, the Company has completed its accounting for all of the tax effects of the Act. Based on the additional analysis performed, no adjustments to the provisional amounts were made in the reporting period which had an impact to the tax provision or consolidated financial statements.
Deferred tax assets and liabilities are recognized for the future tax consequences of differences between the carrying amounts and tax bases of assets and liabilities and operating loss carryforwards and other attributes using enacted rates expected to be in effect when those differences reverse. Valuation allowances are provided against deferred tax assets that are not more likely than not to be realized. Deferred tax assets and liabilities are classified as non-current in the consolidated balance sheet.
The Company provides reserves for potential payments of tax to various tax authorities or does not recognize tax benefits related to uncertain tax positions and other issues. Tax benefits for uncertain tax positions are based on a determination of whether a tax benefit taken by the Company in its tax filings or positions is more likely than not to be realized, assuming that the matter in question will be decided based on its technical merits. The Company’s policy is to record interest and penalties in the provision for income taxes.
The Company regularly assesses its ability to realize deferred tax assets. Changes in historical earnings performance, future earnings projections, and changes in tax laws and tax rates, among other factors, may cause the Company to adjust its valuation allowance on deferred tax assets, which would impact the Company’s income tax expense in the period in which it is determined that these factors have changed.
Excess tax benefits and deficiencies that arise upon vesting or exercise of share-based payments are recognized as an income tax benefit and expense, respectively, in the statement of operations. Excess income tax benefits and are classified as cash flows from operating activities and cash paid to taxing authorities arising from the withholding of shares from employees are classified as cash flows from financing activities.
The Company’s and its subsidiaries’ income tax returns are periodically examined by various tax authorities, including the Internal Revenue Service (IRS) and states. The Company recently completed the audit by the IRS for the years 2013 to 2014 with no material changes to the filed income tax returns. Although the outcome of tax audits is always uncertain and could result in significant cash tax payments, the Company does not believe the outcome of these audits will have a material adverse effect on its consolidated financial position or results of operations.
Loss per Share
Basic net loss per share is determined by dividing net loss by the weighted average shares of common stock outstanding during the period. Diluted net loss per share is determined by dividing net loss by diluted weighted average shares outstanding. Diluted weighted average shares reflects the dilutive effect, if any, of potentially dilutive common shares, such as common stock options calculated using the treasury stock method and convertible notes using the “if-converted” method. In periods with reported net operating losses, all common stock options are deemed anti-dilutive such that basic net loss per share and diluted net loss per share are equal.
The Company’s preferred stock is entitled to receive dividends on an as-if-converted basis in the same form as dividends actually paid on common shares. Accordingly, the preferred stock is considered a participating security and the Company is required to apply the two-class method to consider the impact of the preferred stock on the calculation of basic and diluted earnings per share. The Company is currently in a net loss position and is therefore not required to present the two-class method, however, in the event the Company is in a net income position, the two-class method must be applied by allocating all earnings during the period to common shares and preferred stock based on their contractual entitlements assuming all earnings were distributed.
The calculation of net loss and the number of shares used to compute basic and diluted net loss per share for the years ended December 31, 2018, 2017 and 2016 are as follows:
|
In thousands |
|
2018 |
|
|
2017 |
|
|
2016 |
|
|||
|
Net loss—basic and diluted |
|
$ |
(116,445 |
) |
|
$ |
(67,865 |
) |
|
$ |
(86,350 |
) |
|
Weighted average shares outstanding—basic and diluted |
|
|
297,237 |
|
|
|
270,652 |
|
|
|
211,874 |
|
|
Net loss per share—basic and diluted |
|
$ |
(0.39 |
) |
|
$ |
(0.25 |
) |
|
$ |
(0.41 |
) |
F-11
For the years ended December 31, 2018, 2017 and 2016, the following potentially dilutive securities were not included in the computation of net loss per share because the effect would be anti-dilutive:
|
In thousands |
|
2018 |
|
|
2017 |
|
|
2016 |
|
|
|||
|
Stock options |
|
|
19,263 |
|
|
|
24,108 |
|
|
|
21,188 |
|
|
|
Restricted stock and restricted stock units |
|
|
9,633 |
|
|
|
12,006 |
|
|
|
10,143 |
|
|
|
Exchangeable senior notes (if converted) |
|
|
— |
|
|
|
7,716 |
|
|
|
1,714 |
|
|
|
Preferred stock (if converted) |
|
|
28,932 |
|
|
|
32,818 |
|
|
|
32,818 |
|
|
Debt Instruments
Debt instruments are initially recorded at fair value, with coupon interest and amortization of debt issuance discounts recognized in the consolidated statement of operations as interest expense each period in which such instruments are outstanding. The Company records debt issuance costs related to a recognized debt liability in the consolidated balance sheets as a direct deduction from the carrying amount of that debt liability and amortized to interest expense using the effective interest method over the expected term of the related debt. Unamortized debt issuance costs related to the extinguishment of debt are expensed at the time the debt is extinguished and recorded in other (expense) income, net, in the consolidated statements of operations. If the Company issues shares to discharge the liability, the debt obligation is derecognized and common stock and additional paid-in capital are recognized upon the issuance of those shares.
The 2017 Notes could only be settled in ADSs upon conversion. The terms of the 2017 Notes also allowed for repurchase in cash by the Company at the option of the holders as well as redemption by the Company for cash at specified times. The conversion feature in the 2017 Notes qualified for the exception from derivative accounting in accordance with ASC 815-40 and was therefore accounted for as part of the debt host. The conversion feature in the 2017 Notes was evaluated on a quarterly basis to determine if it still received an exception from derivative accounting in accordance with ASC 815-40. The 2017 Notes were recognized at par of $30.0 million. The Company also recognized a $1.2 million discount related to placement agent fees and offering expenses. This discount was amortized through interest expense through the date of mandatory exchange.
See Note 7—Debt for further discussion.
Stock-Based Compensation
Stock-based compensation cost is generally measured at the grant date, based on the fair value of the award, and is recognized as compensation expense over the requisite service period. For awards with performance conditions, if the achievement of the performance conditions is deemed probable, the Company recognizes compensation expense based on the fair value of the award over the estimated service period. The Company reassesses the probability of achievement of the performance conditions for such awards each reporting period. The Company estimates the level of forfeitures expected to occur based on its historical data and records compensation cost only for those awards that are ultimately expected to vest. See Note 11—Stock Incentive Plans and Stock-Based Compensation for further discussion.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to credit risk consist primarily of cash and cash equivalents and accounts receivable. The Company maintains substantially all of its cash and cash equivalents in financial institutions believed to be of high-credit quality.
A significant portion of the Company’s sales are to wholesalers in the pharmaceutical industry. The Company monitors the creditworthiness of customers to whom it grants credit terms and has not experienced any credit losses. The Company does not require collateral or any other security to support credit sales. Three customers individually accounted for 10% or more of the Company’s gross product sales. Customers A, B, and C accounted for 31%, 30%, and 27%, respectively, of gross product sales for the year ended December 31, 2018 and represented 26%, 24%, and 39%, respectively, of the gross accounts receivable balance as of December 31, 2018. Customers A, B, and C accounted for 27%, 33%, and 28%, respectively, of gross product sales for the year ended December 31, 2017 and represented 27%, 21%, and 41%, respectively, of the gross accounts receivable balance as of December 31, 2017. The Company has not experienced any significant write-offs of its accounts receivable.
Concentration of Suppliers
The Company has contractual freedom to source the API for Vascepa and to procure other services supporting its supply chain and has entered into supply agreements with multiple suppliers. The Company’s supply of product for commercial sale and clinical trials is dependent upon relationships with third-party manufacturers and suppliers.
F-12
The Company cannot provide assurance that its efforts to procure uninterrupted supply of Vascepa to meet market demand will continue to be successful or that it will be able to renew current supply agreements on favorable terms or at all. Significant alteration to or termination of the Company’s current supply chain or its failure to enter into new and similar agreements in a timely fashion, if needed, could have a material adverse effect on its business, condition (financial and other), prospects or results of operations.
The Company currently has manufacturing agreements with multiple independent FDA-approved API manufacturers and several independent FDA-approved API encapsulators and packagers for Vascepa manufacturing. Each of these companies has qualified and validated its manufacturing processes and is capable of manufacturing Vascepa. There can be no guarantee that these or other suppliers with which the Company may contract in the future to manufacture Vascepa will remain qualified to do so to its specifications or that these and any future suppliers will have the manufacturing capacity to meet anticipated demand for Vascepa.
Foreign Currency
All subsidiaries use the U.S. dollar as the functional currency. Monetary assets and liabilities denominated in a foreign currency are remeasured into U.S. dollars at period-end exchange rates. Gains and losses from the remeasurement are included in other (expense) income, net in the consolidated statements of operations. For transactions settled during the applicable period, gains and losses are included in other (expense) income, net in the consolidated statements of operations. Certain amounts payable pursuant to supply contracts are denominated in currencies other than the U.S. dollar.
Fair Value of Financial Instruments
The Company provides disclosure of financial assets and financial liabilities that are carried at fair value based on the price that would be received upon sale of an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Fair value measurements may be classified based on the amount of subjectivity associated with the inputs to fair valuation of these assets and liabilities using the following three levels:
Level 1—Inputs are unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date.
Level 2—Inputs include quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (i.e., interest rates, yield curves, etc.) and inputs that are derived principally from or corroborated by observable market data by correlation or other means (market corroborated inputs).
Level 3—Unobservable inputs that reflect the Company’s estimates of the assumptions that market participants would use in pricing the asset or liability. The Company develops these inputs based on the best information available, including its own data.
The following tables present information about the Company’s assets and liabilities as of December 31, 2018 and 2017 that are measured at fair value on a recurring basis and indicate the fair value hierarchy of the valuation techniques the Company utilized to determine such fair value:
|
|
|
December 31, 2018 |
|
|||||||||||||
|
In thousands |
|
Total |
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
||||
|
Asset: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash equivalents—money markets |
|
$ |
9,880 |
|
|
$ |
9,880 |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
December 31, 2017 |
|
|||||||||||||
|
In thousands |
|
Total |
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
||||
|
Asset: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash equivalents—money markets |
|
$ |
9,317 |
|
|
$ |
9,317 |
|
|
$ |
— |
|
|
$ |
— |
|
F-13
The carrying amounts of cash, cash equivalents, accounts payable and accrued liabilities approximate fair value because of their short-term nature. The carrying amounts and the estimated fair values of debt instruments as of December 31, 2018 and 2017 are as follows:
|
|
|
December 31, 2018 |
|
|
December 31, 2017 |
|
||||||||||
|
In thousands |
|
Carrying Value |
|
|
Estimated Fair Value |
|
|
Carrying Value |
|
|
Estimated Fair Value |
|
||||
|
Current portion of long-term debt from royalty-bearing instrument, net of accrued interest |
|
$ |
33,602 |
|
|
|
|
|
|
$ |
21,569 |
|
|
|
|
|
|
Long-term debt from royalty-bearing instrument |
|
|
46,108 |
|
|
|
|
|
|
|
70,834 |
|
|
|
|
|
|
Total long-term debt from royalty-bearing instrument |
|
$ |
79,710 |
|
|
$ |
78,600 |
|
|
$ |
92,403 |
|
|
$ |
88,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2017 Notes |
|
|
— |
|
|
|
— |
|
|
|
28,992 |
|
|
|
38,200 |
|
The estimated fair value of the long-term debt from royalty-bearing instrument is calculated utilizing the same Level 3 inputs utilized in valuing the related derivative liability (see Derivative Liabilities below). The estimated fair value of the 2017 Notes was calculated based on Level 1 quoted bond prices or, in the absence of quoted bond prices, was calculated using a Level 3 binomial model. The carrying value of the long-term debt from royalty-bearing instrument as of both December 31, 2018 and 2017 did not include a debt discount as it had been fully amortized. The carrying value of the 2017 Notes as of December 31, 2018 and December 31, 2017 included a debt discount of nil and $1.0 million, respectively, which was being amortized as non-cash interest expense over the then expected term of the 2017 Notes, through the first optional put date in January 2022.
Derivative Liabilities
Derivative financial liabilities are recorded at fair value, with gains and losses arising for changes in fair value recognized in the consolidated statement of operations at each period end while such instruments are outstanding. If the Company issues shares to discharge the liability, the derivative financial liability is derecognized and common stock and additional paid-in capital are recognized on the issuance of those shares.
Long-Term Debt Redemption Feature
The Company’s December 2012 royalty-bearing instrument financing arrangement (discussed in Note 7—Debt) contains a redemption feature whereby, upon a change of control, the Company would be required to repay $150.0 million, less any previously repaid amount. The Company determined this redemption feature to be an embedded derivative, which is carried at fair value and is classified as Level 3 in the fair value hierarchy due to the use of significant unobservable inputs. The fair value of the embedded derivative was calculated using a probability-weighted model incorporating management estimates of future revenues and for a potential change in control, and by determining the fair value of the debt with and without the change in control provision included.
The difference between the two was determined to be the fair value of the embedded derivative. The fair value of this derivative liability is remeasured at each reporting period, with changes in fair value recognized in the consolidated statement of operations. As of December 31, 2018, the fair value of the derivative was determined to be nil, and the debt was valued by comparing the debt issues of similar companies with (i) remaining terms of between 1.3 and 4.0 years, (ii) coupon rates of between 5.4% and 10.8% and (iii) market yields of between 6.7% and 15.8%. As of December 31, 2017, the fair value of the derivative was determined to be nil based on current assumptions, and the debt was valued by comparing debt issues of similar companies with (i) remaining terms of between 2.3 and 4.3 years, (ii) coupon rates of between 5.8% and 10.8% and (iii) market yields of between 10.2% and 18.4%. As such, the Company recognized no gain on change in fair value of derivative liability for the year ended December 31, 2018 and 2017.
Any changes in the assumptions used to value the derivative liabilities, including the probability of a change in control, could result in a material change to the carrying value of such liabilities.
Segment and Geographical Information
Operating segments are defined as components of an enterprise about which separate financial information is available that is evaluated on a regular basis by the chief operating decision-maker, or decision-making group, in deciding how to allocate resources to an individual segment and in assessing performance of the segment. The Company currently operates in one business segment, which is the development and commercialization of Vascepa. A single management team that reports to the Company’s chief decision-maker, who is the Chief Executive Officer, comprehensively manages the business. Accordingly, the Company does not have separately reportable segments.
Recent Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board, or FASB, and are early adopted by the Company or adopted as of the specified effective date.
F-14
In May 2014, the FASB issued Accounting Standards Update (“ASU”), No. 2014-09, which amends the guidance for accounting for revenue from contracts with customers. This ASU supersedes the revenue recognition requirements in Accounting Standards Codification (“ASC”) Topic 605, Revenue Recognition, and creates a new Topic 606, Revenue from Contracts with Customers. In 2015 and 2016, the FASB issued additional ASUs related to Topic 606 that delayed the effective date of the guidance and clarified various aspects of the new revenue guidance, including principal versus agent considerations, identifying performance obligations, and licensing, and they include other improvements and practical expedients. The Company adopted this standard effective January 1, 2018 using the modified retrospective transition method.
The Company, as a result of adopting Topic 606 on January 1, 2018, has adjusted its opening retained earnings and deferred revenue balances by $0.2 million. The adjustment relates solely to the Company’s licensing revenues and the timing over which certain non-refundable upfront and milestone payments received from Eddingpharm (Asia) Macao Commercial Offshore Limited and HLS Therapeutics Inc. are recognized under Topic 606. No practical expedients associated with the adoption of Topic 606 were applied.
In May 2017, the FASB issued ASU No.2017-09, Compensation—Stock Compensation: Scope of Modification Accounting. The amendments in ASU No. 2017-09 provide guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. The Company adopted this standard effective January 1, 2018 and, in accordance with the ASU, will apply it prospectively. The Company does not expect it to have a material impact on the Company’s consolidated financial statements.
In January 2017, the FASB issued ASU No. 2017-01, Clarifying the Definition of a Business. The standard clarifies the definition of a business when evaluating whether transactions should be accounted for as acquisitions or disposals of assets or businesses. Under the provisions of this new standard, it is expected that more transactions will be accounted for as asset acquisitions rather than business acquisitions. The Company adopted this standard effective January 1, 2018 and, in accordance with the ASU, will apply it prospectively. The Company does not expect it to have a material impact on the Company’s consolidated financial statements.
In October 2016, the FASB issued ASU No. 2016-16, Income Taxes (Topic 740): Intra-Entity Transfers of Assets Other Than Inventory, which requires the recognition of income tax consequences of an intra-entity transfer of an asset, other than inventory, when the transfer occurs. The Company adopted this standard effective January 1, 2018, which had no impact on the Company’s consolidated financial statements.
In August 2016, the FASB issued ASU No. 2016-15, Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments, which is intended to reduce diversity in practice regarding how certain cash receipts and cash payments related to eight specific issues are presented and classified in the statement of cash flows. In November 2016, the FASB issued ASU No. 2016-18, Statement of Cash Flows (Topic 230): Restricted Cash, which requires that the statement of cash flows explain the change during the period in the total of cash, cash equivalents, and amounts generally described as restricted cash or restricted cash equivalents. The Company adopted this standard effective January 1, 2018 and, in accordance with the ASUs, applied them using a retrospective transition method to each period presented. Adoption of these ASUs did not have a material impact on the Company’s consolidated financial statements.
In January 2016, the FASB issued ASU No. 2016-01, Financial Instruments—Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities. The new guidance is intended to improve the recognition and measurement of financial instruments by requiring separate presentation of financial assets and financial liabilities by measurement category and form of financial asset (i.e., securities or loans and receivables) within the balance sheet or the accompanying notes to the financial statements, eliminating the requirement for public business entities to disclose the method(s) and significant assumptions used to estimate the fair value that is required to be disclosed for financial instruments measured at amortized cost within the balance sheet, requiring public business entities to use the exit price notion when measuring the fair value of financial instruments for disclosure purposes, requiring equity investments (except those accounted for under the equity method of accounting, or those that result in consolidation of the investee) to be measured at fair value with changes in fair value recognized in net income, and requiring a reporting organization to present separately in other comprehensive income the portion of the total change in the fair value of a liability resulting from a change in the instrument-specific credit risk (also referred to as “own credit”) when the organization has elected to measure the liability at fair value in accordance with the fair value option for financial instruments, among others. In February 2018, the FASB issued ASU No. 2018-03, Technical Corrections and Improvements to Financial Instruments—Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities, which is intended to clarify certain aspects of the guidance issued in ASU 2016-01. The Company adopted these standards effective January 1, 2018, which had no impact on the Company’s consolidated financial statements.
The Company also considered the following recent accounting pronouncements which were not yet adopted as of December 31, 2018:
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement (Topic 820): Disclosure Framework—Changes to the Disclosure Requirements for Fair Value Measurement, which eliminates, adds and modifies certain disclosure requirements for fair
F-15
value measurements, including eliminating the requirement to disclose the amount of and reasons for transfers between Level 1 and Level 2 of the fair value hierarchy, and requiring disclosure of the range and weighted average used to develop significant unobservable inputs for Level 3 fair value measurements. The new guidance is effective for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. Early adoption, either of the entire standard or only the provisions that eliminate or modify requirements, is permitted. The Company has evaluated the disclosure requirements of this standard and does not expect it to have a material impact on the Company’s consolidated financial statements.
In June 2018, the FASB issued ASU No. 2018-07, Compensation—Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting, which is intended to simplify the accounting for share-based payments to nonemployees by aligning it with the accounting for share-based payments to employees, with certain exceptions. The new guidance is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. Early adoption is permitted. The Company has evaluated the accounting, transition and disclosure requirements of this standard and does not expect it to have a material impact on the Company’s consolidated financial statements.
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842). The new guidance will require lessees to recognize a right-of-use asset and a lease liability for virtually all of their leases (other than leases that meet the definition of a short-term lease). The liability will be equal to the present value of lease payments. The asset will be based on the liability, subject to adjustment, such as for initial direct costs. Under the new guidance, lessor accounting is largely unchanged but certain targeted improvements were made to align, where necessary, lessor accounting with the lessee accounting model and Topic 606, Revenue from Contracts with Customers. The new lease guidance also simplified the accounting for sale and leaseback transactions primarily because lessees must recognize lease assets and lease liabilities and therefore, will no longer be provided with a source of off-balance sheet financing.
In July 2018, the FASB issued ASU No. 2018-10, which provides narrow amendments to ASU No. 2016-02 to clarify how to apply the rate implicit in the lease, impairment of the net investment in the lease, lessee reassessment of lease classification, variable payments that depend on an index or rate and certain transition adjustments. In July 2018, the FASB also issued ASU No. 2018-11, which provides targeted improvements to ASU No. 2016-02 to provide entities the transition option to not apply the standard in the comparative periods presented in the year of adoption. The Company will adopt the new standard effective January 1, 2019 using the modified retrospective transition method and elect to not apply the standard in the comparative periods presented in the year of adoption. The Company’s assessment of adopting ASU No. 2016-02 consisted of a review of contracts, discussions with key stakeholders and a cataloging of potential impacts on its internal controls, policies and operations. The adoption will not have a material impact on the Company’s consolidated financial statements as of the effective date. As further described in Note 17—Subsequent Events, on February 5, 2019 the Company entered into a lease for new office space. The Company is currently evaluating the impact of this lease to the Company’s consolidated financial statements under ASC 842.
The Company believes that the impact of other recently issued but not yet adopted accounting pronouncements will not have a material impact on the Company’s consolidated financial position, results of operations, and cash flows, or do not apply to the Company’s operations.
(3) Intangible Asset
Intangible asset consists of the historical acquisition cost of certain technology rights for Vascepa and has an estimated weighted-average remaining useful life of 11.6 years. The carrying value as of December 31, 2018 and 2017 is as follows:
|
In thousands |
|
December 31, 2018 |
|
|
December 31, 2017 |
|
||
|
Technology rights |
|
$ |
11,624 |
|
|
$ |
11,624 |
|
|
Accumulated amortization |
|
|
(4,144 |
) |
|
|
(3,498 |
) |
|
Intangible asset, net |
|
$ |
7,480 |
|
|
$ |
8,126 |
|
Amortization expense for each of the years ended December 31, 2018 and 2017 was $0.6 million and is included in research and development expense. Estimated future amortization expense, based upon the Company’s intangible asset, as of December 31, 2018 is as follows:
F-16
|
In thousands |
|
|
|
|
|
|
Amount |
|
||
|
2019 |
|
$ |
646 |
|
|
2020 |
|
|
646 |
|
|
2021 |
|
|
646 |
|
|
2022 |
|
|
646 |
|
|
2023 |
|
|
646 |
|
|
Thereafter |
|
|
4,250 |
|
|
Total |
|
$ |
7,480 |
|
(4) Inventory
The Company capitalizes its purchases of saleable inventory of Vascepa from suppliers that have been qualified by the FDA. Inventories as of December 31, 2018 and 2017 consist of the following:
|
In thousands |
|
December 31, 2018 |
|
|
December 31, 2017 |
|
||
|
Raw materials |
|
$ |
14,142 |
|
|
$ |
7,044 |
|
|
Work in process |
|
|
8,590 |
|
|
|
10,844 |
|
|
Finished goods |
|
|
35,357 |
|
|
|
12,372 |
|
|
Total inventory, gross |
|
|
58,089 |
|
|
|
30,260 |
|
|
Inventory cost adjustment |
|
|
(287 |
) |
|
|
— |
|
|
Inventory |
|
|
57,802 |
|
|
|
30,260 |
|
(5) Property, Plant and Equipment
Property, plant and equipment as of December 31, 2018 and 2017 consist of the following:
|
In thousands |
|
December 31, 2018 |
|
|
December 31, 2017 |
|
||
|
Leasehold improvements |
$ |
156 |
|
|
$ |
156 |
|
|
|
Computer equipment |
|
63 |
|
|
|
63 |
|
|
|
Furniture and fixtures |
|
66 |
|
|
|
66 |
|
|
|
Software |
|
617 |
|
|
|
559 |
|
|
|
Property, plant and equipment |
|
|
902 |
|
|
|
844 |
|
|
Accumulated depreciation and amortization |
|
(839 |
) |
|
|
(816 |
) |
|
|
Property, plant and equipment, net |
|
|
63 |
|
|
|
28 |
|
Depreciation expense for the years ended December 31, 2018, 2017, and 2016 was less than $0.1 million, $0.1 million and $0.1 million, respectively.
(6) Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities consist of the following as of December 31, 2018 and 2017:
|
In thousands |
|
December 31, 2018 |
|
|
December 31, 2017 |
|
||
|
Payroll and payroll-related expenses |
$ |
12,170 |
|
|
$ |
8,298 |
|
|
|
Research and development expenses |
|
— |
|
|
|
1,772 |
|
|
|
Sales and marketing accruals |
|
20,003 |
|
|
|
9,230 |
|
|
|
Accrued revenue allowances |
|
44,286 |
|
|
|
34,445 |
|
|
|
All other |
|
7,712 |
|
|
|
5,157 |
|
|
|
Accrued expenses and other current liabilities |
$ |
84,171 |
|
|
$ |
58,902 |
|
|
F-17
Long-Term Debt from Royalty-Bearing Instrument—December 2012 Financing
On December 6, 2012, the Company entered into a Purchase and Sale Agreement with BioPharma Secured Debt Fund II Holdings Cayman LP, or BioPharma. Under this agreement, the Company granted to BioPharma a security interest in future receivables associated with the Vascepa patent rights, in exchange for $100.0 million received at the closing of the agreement which occurred in December 2012. Under these terms, the Company continues to own all Vascepa intellectual property rights, however, such rights, as described below, could be used as collateral for repayment of the remaining unpaid balance under this agreement if the Company defaults on making required payments. In the agreement, the Company agreed to repay BioPharma up to $150.0 million with such repayment based on a portion of net revenues and receivables generated from Vascepa. On December 20, 2017, BioPharma assigned all rights under this agreement to CPPIB Credit Europe S.à r.l., or CPPIB.
As of December 31, 2018, the remaining amount to be repaid to CPPIB is $88.6 million. During the year ended December 31, 2018, the Company made repayments under the agreement of $20.5 million to CPPIB and an additional $7.7 million was paid in February 2019 for the fourth quarter of 2018. These payments, as well as additional quarterly repayments scheduled in the future, are calculated as 10% of Vascepa net revenues. All such payments reduce the remainder of the $150.0 million in aggregate payments to CPPIB. Except upon a change of control in Amarin, the agreement does not expire until $150.0 million in aggregate has been repaid. The Company can prepay the net remaining amount at any time.
The Company determined the redemption feature upon a change of control to be an embedded derivative requiring bifurcation. The fair value of the embedded derivative was calculated by determining the fair value of the debt with the change in control provision included and also without the change in control provision. The difference between the two fair values of the debt was determined to be the fair value of the embedded derivative, and upon closing the Company recorded a derivative liability of $14.6 million as a reduction to the note payable. The fair value of this derivative liability is remeasured at each reporting period, with changes in fair value recognized in the consolidated statement of operations and any changes in the assumptions used in measuring the fair value of the derivative liability could result in a material increase or decrease in its carrying value. Based on current assumptions underlying the valuation, the Company recognized no gain or loss on change in fair value of derivative liability during the years ended December 31, 2018 and 2017.
As of December 31, 2018 and 2017, the carrying value of the royalty-bearing instrument, net of the unamortized debt discount and issuance costs, was $79.7 million and $92.4 million, respectively. During the year ended December 31, 2018, the Company recorded cash and non-cash interest expense of $5.6 million and $2.0 million, respectively, in connection with the royalty-bearing instrument. During the year ended December 31, 2017, the Company recorded $6.4 million and $2.1 million of cash and non-cash interest expense, respectively, in connection with the royalty-bearing instrument. The Company will periodically evaluate the remaining term of the agreement and the effective interest rate is recalculated each period based on the Company’s most current estimate of repayment.
To secure the obligations under the agreement, the Company granted BioPharma, which it subsequently assigned to CPPIB, a security interest in the Company’s patents, trademarks, trade names, domain names, copyrights, know-how and regulatory approvals related to the covered products, all books and records relating to the foregoing and all proceeds of the foregoing, referred to collectively as the collateral. If the Company (i) fails to deliver a payment when due and does not remedy that failure within a specific notice period, (ii) fails to maintain a first-priority perfected security interest in the collateral in the United States and does not remedy that failure after receiving notice of such failure or (iii) becomes subject to an event of bankruptcy, then CPPIB may attempt to collect the maximum amount payable by the Company under this agreement (after deducting any payments the Company has already made).
Under the agreement, the Company is restricted from paying dividends on its common shares, unless it has cash and cash equivalents in excess of a specified amount after such payment.
January 2012, May 2014, and November 2015 Exchangeable Senior Notes
In 2012, 2014 and 2015, the Company and its subsidiaries entered into a series of transactions pertaining to exchangeable notes. In January 2017, holders of the 3.5% exchangeable senior notes due 2032 (the “2012 Notes”) exercised their option to put approximately $15.0 million in aggregate principal amount of 2012 Notes to the Company for cash and, in March 2017, the Company redeemed the entirety of the remaining $0.1 million in aggregate principal amount of 2012 Notes. The carrying value of the related conversion option will remain in equity hereafter as a result of the repayment in full of the related debt instrument. As of December 31, 2018 and 2017, all debt issued in these transactions was exchanged or redeemed such that none remained outstanding.
January 2017 Exchangeable Senior Notes
On January 20, 2017, the Company and Corsicanto II DAC (“Corsicanto II”), a designated activity company formed under the laws of Ireland and a wholly owned subsidiary of the Company, entered into separate, privately negotiated purchase agreements with certain investors pursuant to which Corsicanto II issued and sold $30.0 million in aggregate principal amount of 3.5% exchangeable senior
F-18
notes due 2047 (the “2017 Notes”) at an issue price of 100%. The net proceeds from the offering were $28.8 million after deducting placement agent fees and offering expenses payable by the Company. The 2017 Notes were issued pursuant to an Indenture (the “Indenture”) entered into by the Company, Corsicanto II and Wilmington Trust, National Association, as trustee (the “Trustee”). The 2017 Notes were the senior unsecured obligations of Corsicanto II and were guaranteed by the Company. The offering of the 2017 Notes closed on January 25, 2017. In October 2018, Corsicanto II mandatorily exchanged $30.0 million of aggregate principal amount of the 2017 Notes for equity upon satisfaction of specified equity conditions as described below, such that no 2017 Notes remained outstanding as of December 31, 2018. Corsicanto II has no assets, operations, revenues or cash flows other than those related to the issuance, administration and repayment of the 2017 Notes.
The 2017 Notes Indenture contained a provision that allowed the Company to elect, at its option, to cause all or any portion of the 2017 Notes to be mandatorily exchanged in whole or in part at any time prior to the close of business on the business day preceding January 15, 2047 if the Daily VWAP (as defined in the Indenture) equaled or exceeded 130% of the Exchange Price then in effect (which quotient equals approximately $5.05 on the date thereof) for at least 20 VWAP Trading Days (as defined in the Indenture) in any 30 VWAP Trading Day period, and upon satisfaction of other specified equity conditions, including that the ADSs issuable upon exchange of the 2017 Notes be eligible for resale without registration by non-affiliates and listed on The NASDAQ Global Market, its related exchanges or the New York Stock Exchange. In October 2018, Corsicanto II gave notice to the holders of the 2017 Notes that the above described conditions had been satisfied and exercised its option to mandatorily exchange $30.0 million of aggregate principal amount of the 2017 Notes for equity with settlement in November 2018, such that all of the outstanding 2017 Notes were retired. The initial exchange rate was 257.2016 ADSs per $1,000 principal amount of the 2017 Notes (equivalent to an initial exchange price of approximately $3.89 per ADS (the “Exchange Price”)), subject to adjustment in certain circumstances, including, but not limited to, the payment of cash dividends. The initial exchange price for the 2017 Notes represented a premium of approximately 35% over the last reported sale price of $2.88 per share of the Company’s ADSs on The NASDAQ Global Market on January 19, 2017. Upon exchange, the 2017 Notes were settled in ADSs. Consistent with the 2017 Notes Indenture, no adjustment was needed such that the final exchange rate was 257.2016 ADSs per $1,000 of principal amount, resulting in the issuance of 7,716,046 ADSs in exchange for the 2017 Notes.
Upon exchange, the Company settled $30.0 million of aggregate principal amount of the 2017 Notes ($29.2 million in carrying value, net of unamortized issuance costs) and recognized $5.0 million in common stock and $24.4 million in additional paid-in capital. Included within this $24.4 million is $0.3 million of accrued but unpaid interest as of the exchange date deemed satisfied and discharged in full upon delivery of the ADSs consistent with the terms of the notes and ASC 470-20, less $0.1 million of transaction costs.
The 2017 Notes had a stated interest of 3.5% per annum from, and including, January 25, 2017, payable semi-annually in arrears on January 15 and July 15 of each year, beginning on July 15, 2017 and ending upon the 2017 Notes’ maturity date of January 15, 2047, had the notes not been exchanged early. The 2017 Notes Indenture provided holders the option to exchange the 2017 Notes for ADSs at any time after the issuance of the 2017 Notes and prior to the close of business on the second business day immediately preceding January 15, 2047 at the initial exchange rate. If a Fundamental Change (as defined in the 2017 Notes Indenture) had occurred prior to the 2017 Notes being exchanged, holders could have required the Company to repurchase all or part of their 2017 Notes for cash at a Fundamental Change repurchase price equal to 100% of the aggregate principal amount of the 2017 Notes to be repurchased, plus accrued and unpaid interest to, but not including, the Fundamental Change repurchase date.
Corsicanto II agreed to use commercially reasonable efforts to procure the listing of the 2017 Notes on the Global Exchange Market operated under the supervision of the Irish Stock Exchange (or on another recognized stock exchange for the purposes of Section 64 of the Taxes Consolidation Act 1997 of Ireland and within the meaning of Section 1005 ITA 2007 of the United Kingdom) prior to July 15, 2017, which was the first interest payment date for the 2017 Notes.
The 2017 Notes were recorded at par of $30.0 million. In addition, the Company recorded a discount of $1.2 million in placement agent fees and offering expenses. Such costs were presented as a direct deduction from the debt liability on the consolidated balance sheet as of December 31, 2017. This discount was amortized as interest expense over the estimated life of the 2017 Notes, through the first optional put date in January 2022. As of December 31, 2018 and 2017, the carrying value of the 2017 Notes, net of unamortized discount, was nil and $29.0 million, respectively.
Because the conversion option in the 2017 Notes received an exception from derivative accounting and only required gross physical settlement in shares, the embedded option did not require separate accounting and was therefore accounted for as part of the debt host at amortized cost. In addition, the Company determined that the fundamental change redemption feature was clearly and closely related to the debt host in accordance with ASC 815-15 and therefore did not require bifurcation.
During the year ended December 31, 2018, the Company recognized interest expense of $1.1 million related to the 2017 Notes, of which $0.2 million represents non-cash interest and $0.9 million represents contractual coupon interest. During the year ended December 31, 2017, the Company recognized interest expense of $1.2 million related to the 2017 Notes, of which $0.2 million represented non-cash interest and $1.0 million represents contractual coupon interest. As of December 31, 2018 and 2017, the Company had accrued interest of nil and $0.5 million, respectively, related to the 2017 Notes, which is presented as current portion of
F-19
exchangeable senior notes, net of discount, on the consolidated balance sheets. The Company made the contractual interest payments due on the 2017 Notes during the years ended December 31, 2018 and 2017 of $1.1 million and $0.5 million, respectively.
(8) Commitments and Contingencies
Litigation
On February 22, 2019, a purported investor in the Company’s publicly traded securities filed a putative class action lawsuit against Amarin Corporation plc, the Company’s chief executive officer and chief scientific officer in the U.S. District Court for the District of New Jersey, Debendra Sharma v. Amarin Corporation plc, John F. Thero and Steven Ketchum, No. 2:19-cv-06601 (D.N.J. Feb. 22, 2019). The lawsuit alleges that, during the period September 24, 2018 and November 9, 2018, the Company misled investors by purportedly not disclosing that the placebo given to patients in the REDUCE-IT study, mineral oil, may have caused cardiovascular problems in the patients taking it, thereby misleading investors on the outcome of the REDUCE-IT study and artificially inflating the price of the Company’s securities. Based on these allegations, the suit asserts claims under the Securities Exchange Act of 1934 and seeks unspecified monetary damages and attorneys’ fees and costs. The Company believes that it has valid defenses and will vigorously defend against the claims, but cannot predict the outcome of this lawsuit. The Company is unable to reasonably estimate the loss exposure, if any, associated with the claims. The Company has insurance coverage that is anticipated to cover any significant loss exposure that may arise from this action after payment by the Company of the associated deductible obligation under such insurance coverage.
On August 30, 2017, Amarin Pharma, Inc. and Amarin Pharmaceuticals Ireland Limited, each wholly-owned subsidiaries of Amarin Corporation plc, filed a lawsuit with the United States International Trade Commission, or the ITC, captioned In the Matter of Certain Synthetically Produced, Predominantly EPA Omega-3 Products in Ethyl Ester or Re-esterified Triglyceride Form, USITC Docket 337-3247, against manufacturers, importers, and distributors of products containing synthetically produced omega-3 products in ethyl ester or re-esterified triglyceride form that contain more EPA than DHA or any other single component for use in or as dietary supplements. The lawsuit sought an investigation by the ITC under Section 337 of the Tariff Act of 1930 (19 U.S.C. §1337), which makes unlawful unfair methods of competition and unfair acts involving the importation and sale of articles in the United States that injure or threaten injury to a domestic industry. On October 27, 2017, the ITC determined to not institute our requested investigation. On December 1, 2017, the Company appealed the ITC’s non-institution decision to the United States Court of Appeals for the Federal Circuit (Case Nos. 18-1247, 18-114). That appeal is ongoing. The Company intends to pursue this matter vigorously.
In September and October 2016, the Company received paragraph IV certification notices from four companies contending to varying degrees that certain of its patents are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of Vascepa as described in those companies’ abbreviated new drug applications, or ANDAs. The Company filed patent infringement lawsuits against three of these four ANDA applicants. In October 2016, Amarin filed a lawsuit against Roxane Laboratories, Inc. and related parties (collectively, “Roxane”) in the U.S. District Court for the District of Nevada. The case against Roxane is captioned Amarin Pharma, Inc. et al. v. Roxane Laboratories, Inc. et al., Civ. A. No. 2:16-cv-02525 (D. Nev.). According to a stipulation filed with the Nevada court, in December 2016, Roxane transferred its ANDA to West-Ward Pharmaceuticals International Limited, which then designated West-Ward Pharmaceuticals Corp. (or together with West-Ward Pharmaceuticals International Limited, West-Ward) as its agent for FDA communications. In view of the ANDA transfer, in February 2017, West-Ward replaced Roxane and related parties as Defendants in the above-referenced case. The case against West-Ward is now captioned Amarin Pharma, Inc. et al. v. West-Ward Pharmaceuticals Corp. et al., Civ. A. No. 2:16-cv-02525 (D. Nev.). In November 2016, Amarin filed a lawsuit against Dr. Reddy’s Laboratories, Inc. and Dr. Reddy’s Laboratories, Ltd. (collectively, “DRL”) in the U.S. District Court for the District of Nevada. The case against DRL is captioned Amarin Pharma, Inc. et al. v. Dr. Reddy’s Laboratories, Inc. et al., Civ. A. No. 2:16-cv-02562 (D. Nev.). In November 2016, Amarin filed a lawsuit against Teva Pharmaceuticals USA, Inc. and Teva Pharmaceuticals Industries Limited (collectively, “Teva”) in the U.S. District Court for the District of Nevada. The case against Teva is captioned Amarin Pharma, Inc. et al. v. Teva Pharmaceuticals USA, Inc. et al., Civ. A. No. 2:16-cv-02658. In all three lawsuits, Amarin is seeking, among other remedies, an order enjoining each defendant from marketing generic versions of Vascepa before the last to expire of the asserted patents in 2030. The three lawsuits have been consolidated for pretrial proceedings. As a result of the statutory stay associated with the filing of these lawsuits under the Hatch-Waxman Act, the FDA cannot grant final approval to West-Ward, DRL, or Teva’s respective ANDA before January 2020, unless there is an earlier court decision holding that the subject patents are not infringed and/or are invalid.
The fourth ANDA applicant referenced above is Apotex Inc. (“Apotex”), which sent Amarin a paragraph IV certification notice in September 2016. The notice reflected that Apotex made a paragraph IV notice as to some, but not all, of the patents listed in the Orange Book for Vascepa. Because Apotex did not make a paragraph IV certification as to all listed patents, Apotex cannot market a generic version of Vascepa before the last to expire of the patents for which Apotex did not make a paragraph IV certification, which is in 2030. At a later date, Apotex may elect to amend its ANDA in order to make a paragraph IV certification as to additional listed patents. If and when Apotex does make such an amendment, it would be required to send Amarin an additional paragraph IV certification notice, and Amarin would then have the ability to file a lawsuit against Apotex pursuant to the Hatch-Waxman Act.
F-20
In October 2016, the Company introduced to the market a 0.5-gram dose strength of Vascepa. In August 2017, as anticipated, the Company received a paragraph IV certification notice from Teva contending that certain of its patents are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of the 0.5-gram dose strength of Vascepa, as described in the Teva ANDA. This Teva ANDA was filed as an amendment to the 1-gram Teva ANDA and is related to patents already at issue in the 1-gram Vascepa patent litigation. This certification followed the related listing in the Orange Book of patents associated with the 0.5-gram product in June 2017. This June 2017 listing was within the five-year, post NDA-approval period during which the Hatch-Waxman Amendments require a paragraph IV certification of patent invalidity or non-infringement under the Hatch-Waxman, five-year, NCE regulatory scheme. Accordingly, in October 2017, the Company filed a patent infringement lawsuit against Teva in the U.S. District Court for the District of Nevada. The case is captioned Amarin Pharma, Inc. et al. v. Teva Pharmaceuticals USA, Inc. et al., Civ. A. No. 2:17-cv-2641 (D. Nev.). In this lawsuit, the Company sought, among other remedies, an order enjoining Teva from marketing generic versions of the 0.5-gram dose strength of Vascepa before the last to expire of the asserted patents in 2030.
On May 24, 2018, the Company entered into a settlement agreement with Teva that resolves its ANDA patent litigation as it relates to Teva’s as amended ANDA for both the 1-gram and 0.5-gram dose strengths of Vascepa. As part of this settlement agreement, Teva may first begin selling its generic version of Vascepa in the United States on August 9, 2029, or earlier under certain customary circumstances, including commercial launch by another generic manufacturer under certain circumstances, in which event Teva would pay the Company certain royalties on its generic Vascepa products. The ANDA patent litigation continues in the United States District Court for the District of Nevada with parties West-Ward and DRL.
In July 2018, as anticipated, the Company received a paragraph IV certification notice from DRL contending that certain of its patents are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of the 0.5-gram dose strength of Vascepa, as described in the DRL ANDA. This DRL ANDA was filed as an amendment to the 1-gram DRL ANADA and is related to patents already at issue in the 1-gram Vascepa patent litigation. This certification followed the related listing in the Orange Book of patents associated with the 0.5-gram product in June 2017. This June 2017 listing was within the five-year, post NDA-approval period during which the Hatch-Waxman Amendments require a paragraph IV certification of patent invalidity or non-infringement lawsuit against DRL in the U.S. District Court for the District of Nevada. The case is captioned Amarin Pharma, Inc. et al. v. Dr. Reddy’s Laboratories, Inc. et al., Civ. A. No. 2:18-cv-01596 (D. Nev.). In this lawsuit, the Company is seeking, among other remedies, an order enjoining DRL from marketing generic versions of the 0.5-gram dose strength of Vascepa before the last to expire of the asserted patents in 2030. In light of the overlap between the cases, DRL and Amarin have stipulated that the final judgment on the merits of the parties’ contentions in the consolidated 1-gram action shall also be binding in the 0.5-gram case.
The Company intends to vigorously enforce its intellectual property rights relating to Vascepa, but cannot predict the outcome of these lawsuits or any subsequently filed lawsuits.
In addition to the above, in the ordinary course of business, the Company is from time to time involved in lawsuits, claims, investigations, proceedings, and threats of litigation relating to intellectual property, commercial arrangements and other matters.
Leases
The Company leases office space under operating leases. Future minimum lease payments under these leases as of December 31, 2018 are as follows:
|
In thousands |
|
|
|
|
|
Year Ending December 31, |
|
Operating Leases |
|
|
|
2019 |
|
$ |
200 |
|
|
2020 |
|
|
— |
|
|
2021-2023 |
|
|
— |
|
|
Total |
|
$ |
200 |
|
On September 30, 2011, the Company entered into an agreement for 320 square feet of office space at 2 Pembroke House, Upper Pembroke Street 28-32 in Dublin, Ireland. The office space was subsequently reduced to 270 square feet, effective November 1, 2013. The agreement began November 1, 2011 and terminates on October 31, 2019 and can be extended automatically for successive one-year periods. Monthly rent is approximately €4,200 (approximately $4,900 at the time of filing). The agreement can be terminated by either party with three months prior written notice.
On July 1, 2011, the Company leased 9,747 square feet of office space in Bedminster, New Jersey. On December 6, 2011 the Company leased an additional 2,142 square feet of space in the same location. On December 15, 2012 and May 8, 2013, the Company leased an additional 2,601 and 10,883 square feet of space, respectively, in the same location. In January 2014 and April 2014, the
F-21
Company entered into separate transactions with the landlord of this property to vacate approximately 2,142 and 2,000 square feet of space in exchange for discounts on contractual future rent payments. In January 2015, the Company executed an agreement to sublease approximately 4,700 square feet of this property to a third party, effective April 1, 2015. This sublease agreement was terminated as of September 30, 2017. Additionally, in June 2015, the Company executed an agreement to sublease approximately 2,500 square feet of this property to a separate third party, effective June 16, 2015, which agreement naturally ceased on March 31, 2018. On December 15, 2016, the Company leased an additional 732 square feet of space in the same location, effective January 1, 2017. The lease, as amended, terminates on April 30, 2019, but the Company is currently in the process of extending this lease through the start of the new Bridgewater, New Jersey lease, as described below. On January 26, 2019, the Company leased an additional 5,988 square feet in an annex building, effective February 1, 2019 and terminating June 30, 2019, which will result in additional rent of approximately $14,000 per month during that period.
As described more fully in Note 17—Subsequent Events, given the anticipated expiration in 2019 of the lease described above, on February 5, 2019, the Company entered into a lease agreement for approximately 67,747 square feet of office space in Bridgewater, New Jersey with a commencement date anticipated to be on or about July 1, 2019 and initial term of 11 years. Under the lease, the Company will pay monthly rent of approximately $141,000 for the first year following the commencement date, with nominal percentage increases every year following.
Total rent expense during the years ended 2018, 2017 and 2016 was approximately $0.8 million, $0.6 million, and $0.6 million, respectively.
Milestone and Supply Purchase Obligations
The Company entered into long-term supply agreements with multiple FDA-approved API suppliers and encapsulators. Certain supply agreements require annual minimum volume commitments by the Company and certain volume shortfalls may require payments for such shortfalls.
These agreements included requirements for the suppliers to meet certain product specifications and qualify their materials and facilities with applicable regulatory authorities including the FDA. The Company has incurred certain costs associated with the qualification of product produced by these suppliers.
Pursuant to the supply agreements, there is a total of $53.0 million that is potentially payable over the term of such agreements based on minimum purchase obligations. The Company continues to meet its contractual purchase obligations.
Under the 2004 share repurchase agreement with Laxdale Limited (“Laxdale”), upon receipt of marketing approval in Europe for the first indication for Vascepa (or first indication of any product containing Amarin Neuroscience Limited intellectual property acquired from Laxdale in 2004), the Company must make an aggregate stock or cash payment to the former shareholders of Laxdale (at the sole option of each of the sellers) of £7.5 million (approximately $9.6 million as of December 31, 2018). Also under the Laxdale agreement, upon receipt of a marketing approval in the United States or Europe for a further indication of Vascepa (or further indication of any other product using Amarin Neuroscience Limited intellectual property), the Company must make an aggregate stock or cash payment (at the sole option of each of the sellers) of £5 million (approximately $6.4 million as of December 31, 2018) for each of the two potential market approvals (i.e., £10 million maximum, or approximately $12.7 million as of December 31, 2018).
The Company has no provision for any of the obligations above since the amounts are either not probable or able to be estimated as of December 31, 2018.
(9) Equity
Preferred Stock
On March 5, 2015, the Company entered into a subscription agreement with four institutional investors (the “Purchasers”), including both existing and new investors, for the private placement of 352,150,790 restricted American Depositary Shares, each representing one (1) share of Amarin’s Series A Convertible Preference Shares, par value £0.05 per share, in the capital of the Company (“Series A Preference Shares”), resulting in gross proceeds to the Company of $52.8 million. The closing of the private placement occurred on March 30, 2015.
For each restricted American Depositary Share, the Purchasers paid a negotiated price of $0.15 (equating to $1.50 on an as-if-converted-to-ordinary-shares basis), resulting in $52.8 million in aggregate gross proceeds to the Company, before deducting estimated offering expenses of approximately $0.7 million. The net proceeds are reflected as preferred stock in the accompanying consolidated balance sheets.
Each ten (10) Series A Preference Shares may be consolidated and redesignated as one (1) ordinary share, par value £0.50 per share, in the capital of the Company, each ordinary share to be represented by American Depositary Shares (“ADSs”), provided that consolidation will be prohibited if, as a result, the holder of such Series A Preference Shares and its affiliates would beneficially own
F-22
more than 4.99% of the total number of Amarin ordinary shares or ADSs outstanding following such redesignation (the “Beneficial Ownership Limitation”). By written notice to the Company, a holder may from time to time increase or decrease the Beneficial Ownership Limitation to any other percentage not in excess of 19.9% specified in such notice; provided that any such increase will not be effective until the sixty-first (61st) day after such notice is delivered to the Company. This consolidation and redesignation may be effected by a holder of Series A Preference Shares following the first to occur of the resale of the ADSs representing the ordinary shares being registered for resale under the Securities Act pursuant to an effective registration statement, following any sale of the ADSs representing the ordinary shares pursuant to Rule 144 under the Securities Act, or if such ADSs representing the ordinary shares are eligible for sale under Rule 144, following the expiration of the one-year holding requirement under Rule 144. During the year ended December 31, 2015, at the request of the holders, a portion of the Series A Preference Shares were consolidated and redesignated, resulting in the issuance of 6,283,333 ADSs.
Except as otherwise provided in the Series A Preference Share Terms or as required by applicable law, the Series A Preference Shares have no voting rights. However, as long as any Series A Preference Shares are outstanding, the Company cannot, without the approval of the holders of seventy-five percent (75%) of the then outstanding Series A Preference Shares, alter or change adversely the powers, preferences or rights attaching to the Series A Preference Shares or enter into any agreement with respect to the foregoing.
Holders of the Series A Preference Shares are entitled to receive, and the Company is required to pay, dividends (other than dividends in the form of ordinary shares) on the Series A Preference Shares equal (on an as-if-converted-to-ordinary-shares basis) to and in the same form as dividends (other than dividends in the form of ordinary shares) actually paid on ordinary shares when, as and if such dividends (other than dividends in the form of ordinary shares) are paid on the ordinary shares.
The restricted American Depositary Shares and Series A Preference Shares were sold in a transaction exempt from the registration requirements under the Securities Act of 1933, as amended (the “Securities Act”). The Company filed a registration statement with the SEC covering the resale of the restricted American Depositary Shares and the ADSs representing ordinary shares created by the consolidation and redesignation of the Series A Preference Shares (the “Registrable Securities”) on April 9, 2015, which was declared effective by the SEC on May 1, 2015. In addition, the Company agreed to use its commercially reasonable best efforts to keep the registration, and any qualification, exemption or compliance under state securities laws which the Company determines to obtain, continuously effective, and to keep the Registration Statement free of any material misstatements or omissions, until the earlier of (a) March 11, 2017 or (b) the date on which all Registrable Securities held by Purchasers may be sold or transferred in compliance with Rule 144 under the Securities Act, without any volume or manner of sale restrictions.
The Series A Preference Shares contain a contingent beneficial conversion feature (“BCF”) because they contain a conversion feature at a fixed rate that was in-the-money when issued. The BCF was recorded in the three months ended June 30, 2015 as a result of the related Form S-3 Registration Statement being declared effective, which represents the resolution of the contingency to convert the Series A Preference Shares. The BCF was recognized in stockholders’ deficit and was measured by allocating a portion of the proceeds equal to the intrinsic value of that feature to additional paid-in capital. The effective purchase price of the ordinary shares into which the preferred shares are convertible was $1.50, which was used to compute the intrinsic value. The intrinsic value was calculated as the difference between the effective purchase price of the ordinary shares and the market value ($2.39 per share) on the date the preferred shares were issued, multiplied by the number of shares into which the preferred shares are convertible. The BCF resulting from the issuance of the Series A Preference Shares was determined to be $31.3 million. The BCF was recorded as a non-cash dividend to preferred shareholders through accumulated deficit, and was therefore reflected as an adjustment to net loss applicable to common shareholders for earnings per common share purposes in accordance with GAAP for the year ended December 31, 2015.
On March 30, 2015, in connection with the closing of the private placement, and pursuant to a pre-existing contractual right to participate in certain private placement transactions effected by the Company, the Company entered into a separate subscription agreement with an existing investor, Sofinnova Venture Partners VII L.P. (Sofinnova), for the purchase of an additional $5.8 million of restricted American Depositary Shares, each representing one (1) share of the Company’s Series A Preference Shares, at the same price per share and otherwise on substantially the same terms as the initial private placement (the “Second Private Placement”). In accordance with applicable marketplace rules of the NASDAQ Stock Market, the consummation of the Second Private Placement was conditioned upon approval by the Company’s shareholders at a future meeting of the Company’s shareholders. Such approval was received at the Company’s Annual General Meeting of Shareholders on July 6, 2015 and as a result, the closing of the Second Private Placement occurred on July 10, 2015. The Company issued 38,867,180 restricted ADSs, each representing one Series A Preference Share, which could be consolidated and redesignated from time to time up to a maximum of 3,886,718 ordinary shares, each ordinary share to be represented by one ADS. For each restricted ADS, Sofinnova paid a negotiated price of $0.15 (equating to $1.50 on an as-if-converted-to-ordinary-shares basis) resulting in gross proceeds to the Company of $5.8 million.
The Company filed another registration statement with the SEC covering the resale of these restricted American Depositary Shares and the ADSs representing ordinary shares created by the consolidation and redesignation of the Series A Preference Shares (the “Sofinnova Registrable Securities”) on July 24, 2015, which was declared effective by the SEC on August 7, 2015. In addition, the
F-23
Company agreed to use its commercially reasonable best efforts to keep the registration, and any qualification, exemption or compliance under state securities laws which the Company determines to obtain, continuously effective, and to keep the registration statement free of any material misstatements or omissions, until the earlier of (a) July 10, 2017 or (b) the date on which all Sofinnova Registrable Securities held by Sofinnova may be sold or transferred in compliance with Rule 144 under the Securities Act, without any volume or manner of sale restrictions.
The existence of this preferred stock purchase option was determined to be a derivative liability effective March 5, 2015, the date on which the private placement was initially subscribed. The fair value of this liability was calculated using a Black-Scholes model and was determined to be $0.9 million at inception and was charged to accumulated deficit as a deemed non-cash dividend to Sofinnova. The liability was then marked to fair value as of March 30, 2015, the date on which the Company executed a subscription agreement with Sofinnova, resulting in a charge of $0.9 million through gain (loss) on change in fair value of derivatives. The liability of $1.8 million was reclassified to permanent equity (additional paid-in capital) on such date. Subsequent to approval of the Second Private Placement at the Company’s Annual General Meeting of Shareholders in July 2015, the Company recorded the remaining value of the BCF related to this share issuance as a non-cash dividend to preferred shareholders through accumulated deficit. The value of the BCF was determined on the same basis as the first private placement and amounted to $3.4 million less $1.8 million previously recorded for the preferred stock purchase option for a net non-cash charge of $1.6 million in the year ended December 31, 2015.
During the year ended December 31, 2018, the Company issued 3,886,718 ADSs upon consolidation and redesignation of Series A Preference Shares at the request of the holder, such that a maximum of 28,931,746 ordinary shares remain issuable upon future consolidation and redesignation of the remaining Series A Preference Shares as of December 31, 2018, subject to certain adjustments for dilutive events.
Common Stock
On November 29, 2018, the Company completed a public offering of 11,111,112 ADSs, with each ADS representing one ordinary share of the Company. The underwriters purchased the ADSs from the Company at a price of $17.575 per ADS after commission, resulting in net proceeds to the Company of approximately $194.8 million, after deducting customary commissions and offering expenses. The stated uses of net proceeds in connection with this offering were as follows: to support the ongoing commercialization of Vascepa following REDUCE-IT results, including (i) seeking regulatory approval to expand the approved label for Vascepa, (ii) expansion of the Company’s sales force, and (iii) support of expanded commercial operations, to increase commercial supply of Vascepa from third-party drug product suppliers, and for general corporate purposes.
On February 1, 2018, the Company completed a public offering of 19,178,082 ADSs, with each ADS representing one ordinary share of the Company. The Company also granted the underwriters a 30-day option to purchase an additional 2,876,712 ADSs, which was partially exercised on March 5, 2018 for issuance of 1,438,356 ADSs. The underwriters purchased the ADSs from the Company at a price of $3.41 per ADS after commission, resulting in net proceeds to the Company of approximately $70.0 million, after deducting customary commissions and offering expenses. The stated uses of net proceeds in connection with this offering were as follows: to expand medical education and market awareness initiatives, including, in advance of REDUCE-IT results being known, pilot testing of new promotional initiatives for potential broader application following REDUCE-IT results, to increase its inventory balances for incremental inventory build prior to REDUCE-IT results and for general corporate and working capital purposes.
Incentive Equity Awards
As of December 31, 2018, there were an aggregate of 19,263,038 stock options and 9,633,250 restricted stock units (“RSUs”) outstanding, representing approximately 5% and 3%, respectively, of outstanding shares (including common and preferred shares) on a fully diluted basis.
During the years ended December 31, 2018 and 2017, the Company issued 8,138,305 and 356,656 shares, respectively, as a result of the exercise of stock options, resulting in gross and net proceeds of $26.4 million during the year ended December 31, 2018 and $0.6 million during the year ended December 31, 2017.
In September 2018, in connection with positive REDUCE-IT results, the Company issued 2,499,750 shares upon vesting of performance-based RSUs granted in 2015, of which 764,819 shares were retained as treasury shares as settlement of employee tax obligations.
On May 14, 2018, the Company granted a total of 190,034 RSUs and 286,536 stock options to members of the Company’s Board of Directors under the Amarin Corporation plc Stock Incentive Plan (the “2011 Plan”). The RSUs vest in equal installments over a three-year period upon the earlier of the anniversary of the grant date or the Company’s annual general meeting of shareholders in such anniversary year. The stock options vest in full upon the earlier of the one-year anniversary of the grant date or the Company’s annual
F-24
general meeting of shareholders in such anniversary year. Upon termination of service to the Company or upon a change of control, each Director shall be entitled to a payment equal to the fair market value of one share of Amarin common stock per award vested or granted, respectively, which is required to be made in shares.
On February 1, 2018, the Company granted a total of 1,305,575 RSUs and 2,205,075 stock options to employees under the 2011 Plan. The RSUs vest annually over a three-year period and the stock options vest monthly over a four-year period.
On May 15, 2017, the Company granted a total of 91,504 RSUs and 131,575 stock options to members of the Company’s Board of Directors under the 2011 Plan. The RSUs vest in equal installments over a three-year period upon the earlier of the anniversary of the grant date or the Company’s annual general meeting of shareholders in such anniversary year. The stock options vest in full upon the earlier of the one-year anniversary of the grant date or the Company’s annual general meeting of shareholders in such anniversary year. Upon termination of service to the Company or upon a change of control, each Director shall be entitled to a payment equal to the fair market value of one share of Amarin common stock per award vested or granted, respectively, which is required to be made in shares.
On May 15, 2017, October 2, 2017, March 12, 2018, and November 1, 2018, the Company granted a total of 2,310,000 RSUs, 220,000 RSUs, 970,000 RSUs, and 90,000 RSUs, respectively, to employees under the 2011 Plan that vest over three years commencing after REDUCE-IT results upon the achievement of certain regulatory and sales performance conditions associated with the REDUCE-IT clinical trial and subsequent revenue growth.
On February 1, 2017, the Company granted a total of 1,575,000 RSUs and 2,642,500 stock options to employees under the 2011 Plan. The RSUs vest annually over a three-year period and the stock options vest over a four-year period. During the year ended December 31, 2018, the Company issued 506,679 common shares related to the vesting of these RSUs, of which 183,828 shares were retained as treasury shares as settlement of employee tax obligations.
See Note 11—Stock Incentive Plans and Stock Based Compensation for further information regarding the Company’s incentive equity awards.
(10) Income Taxes
Interest and penalties related to any uncertain tax positions have historically been insignificant. The Company recognizes interest and penalties related to uncertain tax positions within the provision for income taxes. The total amount of unrecognized tax benefits that would affect the Company’s effective tax rate if recognized is nil as of both December 31, 2018 and 2017.
The following is a reconciliation of the total amounts of unrecognized tax benefits for the years ended December 31, 2018, 2017 and 2016:
|
|
|
2018 |
|
|
2017 |
|
|
2016 |
|
||||
|
Beginning uncertain tax benefits |
|
$ |
1,734 |
|
|
$ |
1,633 |
|
|
$ |
1,550 |
|
|
|
Prior year—increases |
|
|
296 |
|
|
|
— |
|
|
|
— |
|
|
|
Prior year—decreases |
|
|
(762 |
) |
|
|
(20 |
) |
|
|
— |
|
|
|
Current year—increases |
|
|
5,547 |
|
|
|
121 |
|
|
|
83 |
|
|
|
Ending uncertain tax benefits |
|
$ |
6,815 |
|
|
$ |
1,734 |
|
|
$ |
1,633 |
|
|
The Company files income tax returns in the United States, Ireland and United Kingdom, or UK. The Company remains subject to tax examinations in the following jurisdictions as of December 31, 2018:
|
Jurisdiction |
|
|
Tax Years |
|
|
2015-2018 |
||
|
United States—State |
|
2012-2018 |
|
|
Ireland |
|
2014-2018 |
|
|
United Kingdom |
|
2017-2018 |
|
The Company does not expect any gross liabilities to expire in 2019 based on statutory lapses or audits.
F-25
The components of loss from operations before taxes were as follows for the years ended December 31, 2018, 2017 and 2016:
|
|
2018 |
|
|
2017 |
|
|
2016 |
|
|||||
|
United States |
|
$ |
(13,583 |
) |
|
$ |
(2,075 |
) |
|
$ |
(8,115 |
) |
|
|
Ireland and United Kingdom |
|
|
(102,766 |
) |
|
|
(52,743 |
) |
|
|
(68,266 |
) |
|
|
|
|
|
$ |
(116,349 |
) |
|
$ |
(54,818 |
) |
|
$ |
(76,381 |
) |
The provision for income taxes shown in the accompanying consolidated statements of operations consists of the following for fiscal 2018, 2017 and 2016:
|
In thousands |
|
2018 |
|
|
2017 |
|
|
2016 |
|
|||
|
Current: |
|
|
|
|
|
|
|
|
|
|
|
|
|
United States—Federal |
|
$ |
4 |
|
|
$ |
1,769 |
|
|
$ |
1,033 |
|
|
United States—State |
|
|
92 |
|
|
|
196 |
|
|
|
138 |
|
|
Total current |
|
$ |
96 |
|
|
$ |
1,965 |
|
|
$ |
1,171 |
|
|
Deferred: |
|
|
|
|
|
|
|
|
|
|
|
|
|
United States—Federal |
|
|
(1,968 |
) |
|
|
5,760 |
|
|
|
(4,001 |
) |
|
United States—State |
|
|
(1,325 |
) |
|
|
(487 |
) |
|
|
(334 |
) |
|
Ireland and United Kingdom |
|
|
(5,435 |
) |
|
|
(16,306 |
) |
|
|
(143 |
) |
|
Change in valuation allowance |
|
|
8,728 |
|
|
|
22,115 |
|
|
|
13,276 |
|
|
Total deferred |
|
$ |
— |
|
|
$ |
11,082 |
|
|
$ |
8,798 |
|
|
Provision for income taxes |
|
$ |
96 |
|
|
$ |
13,047 |
|
|
$ |
9,969 |
|
The provision for income taxes differs from the amount computed by applying the statutory income tax rate to income before taxes due to the following for fiscal 2018, 2017 and 2016:
|
In thousands |
|
2018 |
|
|
2017 |
|
|
2016 |
|
|||
|
Benefits from taxes at statutory rate |
$ |
(29,087 |
) |
|
$ |
(13,698 |
) |
|
$ |
(19,039 |
) |
|
|
Rate differential |
|
9,796 |
|
|
|
3,071 |
|
|
|
4,667 |
|
|
|
Change in valuation reserves |
|
8,728 |
|
|
|
22,115 |
|
|
|
13,276 |
|
|
|
Derivative liabilities |
|
337 |
|
|
|
— |
|
|
|
(668 |
) |
|
|
Nondeductible employee compensation |
|
3,058 |
|
|
|
1,668 |
|
|
|
1,164 |
|
|
|
Stock option/RSU windfall |
|
(7,684 |
) |
|
|
(1,182 |
) |
|
|
— |
|
|
|
Research and development credits |
|
(1,438 |
) |
|
|
(1,177 |
) |
|
|
(1,689 |
) |
|
|
Tax return to provision adjustments |
|
6,736 |
|
|
|
5,788 |
|
|
|
4,524 |
|
|
|
U.S. rate change—tax reform |
|
— |
|
|
|
7,398 |
|
|
|
— |
|
|
|
Cumulative translation adjustment |
|
5,711 |
|
|
|
(12,554 |
) |
|
|
7,385 |
|
|
|
Permanent and other |
|
(404 |
) |
|
|
1,635 |
|
|
|
(1,573 |
) |
|
|
Non-deductible interest expense |
|
267 |
|
|
|
(17 |
) |
|
|
1,922 |
|
|
|
Tax reserves |
|
4,956 |
|
|
|
— |
|
|
|
— |
|
|
|
Long-term debt from royalty-bearing instrument |
|
(880 |
) |
|
|
— |
|
|
|
— |
|
|
|
Provision for income taxes |
$ |
96 |
|
|
$ |
13,047 |
|
|
$ |
9,969 |
|
|
The Company is subject to a corporate tax rate in Ireland of 25% for non-trading activities and 12.5% for trading activities. For the years ended December 31, 2018, 2017, and 2016, the Company applied the statutory corporate tax rate of 25% for Amarin Corporation plc, reflecting the non-trading tax rate in Ireland. However, for Amarin Pharmaceuticals Ireland Limited, a wholly-owned subsidiary of Amarin Corporation plc, the Company applied the 12.5% Irish trading tax rate. In the table above, the Company used Amarin Corporation plc’s 25% tax rate as the starting point for the reconciliation since it is the parent entity of the business.
On December 22, 2017, the U.S. enacted the Act that instituted fundamental changes to the taxation of multinational corporations. The Act includes changes to the taxation of foreign earnings by implementing a dividend exemption system, expansion of the current anti-deferral rules, a minimum tax on low-taxed foreign earnings and new measures to deter base erosion. The Act also includes a permanent reduction in the corporate tax rate to 21%, repeal of the corporate alternative minimum tax, expensing of capital investment, and limitation of the deduction of interest expense. Furthermore, as part of the transition to the new tax system, a one-time transition tax is imposed on a U.S. shareholder's historical undistributed earnings of foreign affiliates. Although the Act was generally
F-26
effective January 1, 2018, U.S. GAAP required recognition of the tax effects of new legislation during the reporting period that included the enactment date, which was December 22, 2017.
As a result of the financial reporting implications of the Act, the SEC provided guidance that allowed the Company to record provisional amounts for those impacts, with the requirement that the accounting be completed in a period not to exceed one year from the date of enactment. As of December 31, 2018, the Company has finalized the amounts related to tax reform to account for the impact of the Act. No adjustments to the financial statements were recorded in connection with the finalization of the accounting.
The primary impact of the Act on the Company related to the re-measurement of deferred tax assets and liabilities resulting from the change in the corporate tax rate from 34% to 21%. At the date of enactment, the Company had net deferred tax assets for the excess of the net tax value over the book basis of its U.S. assets and liabilities which will generate future tax deductions in excess of book expense. As a result of the Act, future tax deductions will result in a decreased reduction in tax expense. Consequently, the Company reduced the amount of the U.S. subsidiary’s net deferred tax assets as of the date of enactment and recorded a non-cash charge of $2.4 million in the provision for income taxes for the year ended December 31, 2017 due to the decrease in the corporate tax rate. In addition, based on the Company’s evaluation of available evidence, the Company recognized non-cash tax expense during the year ended December 31, 2017 of $8.7 million related to the recording of additional valuation allowance to reduce the deferred tax assets on the balance sheet to zero as the Company concluded that it is not more likely than not that certain of the deferred tax benefits resulting from deferred tax assets generated from the U.S. subsidiary operations will be realized.
In April 2016, the Company adopted ASU No. 2016-09, Compensation-Stock Compensation (Topic 718): Improvements to Share-Based Payment Accounting which changes the accounting for certain aspects of share-based payments to employees. One aspect of the standard requires that excess tax benefits and deficiencies that arise upon vesting or exercise of share-based payments be recognized as an income tax benefit and expense in the income statement. Previously, such amounts were recognized as an increase and decrease in additional paid-in capital. This aspect of the standard was adopted prospectively, and accordingly the provisions for income taxes for the years ended December 31, 2018, 2017 and 2016 includes $7.7 million of excess tax benefits, $1.3 million of excess tax benefits and $0.4 million of excess tax deficiencies, respectively, arising from share-based payments during the period of adoption. Additionally, the new standard requires that historical excess tax benefits that were not previously recognized because the related tax deduction had not reduced current taxes should be recognized on a modified retrospective basis as a cumulative-effect adjustment to retained earnings as of the beginning of the annual period of adoption. Consequently, the Company recognized deferred tax assets of approximately $1.6 million relating to excess tax benefits on stock-based compensation during the year of adoption, with a corresponding cumulative-effect adjustment to accumulated deficit.
The income tax effect of each type of temporary difference comprising the net deferred tax asset as of December 31, 2018 and 2017 is as follows:
|
In thousands |
|
December 31, 2018 |
|
|
December 31, 2017 |
|
|||
|
|
|
|
|
|
|
|
|
||
|
Net operating losses |
|
$ |
119,355 |
|
|
$ |
110,715 |
|
|
|
Stock-based compensation |
|
|
8,113 |
|
|
|
12,446 |
|
|
|
Tax credits |
|
|
7,816 |
|
|
|
6,378 |
|
|
|
Other reserves and accrued liabilities |
|
|
6,344 |
|
|
|
3,587 |
|
|
|
Gross deferred tax assets |
|
|
141,628 |
|
|
|
133,126 |
|
|
|
Less: valuation allowance |
|
|
(140,117 |
) |
|
|
(131,389 |
) |
|
|
Total deferred tax assets |
|
|
1,511 |
|
|
|
1,737 |
|
|
|
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
(1,011 |
) |
|
|
(1,145 |
) |
|
|
Other liabilities |
|
|
|
(500 |
) |
|
|
(592 |
) |
|
Total deferred tax liabilities |
|
|
(1,511 |
) |
|
|
(1,737 |
) |
|
|
Net deferred tax assets |
|
$ |
— |
|
|
$ |
— |
|
|
The Company assesses whether it is more-likely-than-not that the Company will realize its deferred tax assets. The Company determined that it was more-likely-than-not that the Irish, U.S., UK, and Israeli net operating losses and the related deferred tax assets would not be realized in future periods and a full valuation allowance has been provided for all periods.
F-27
The following table reflects the activity in the valuation allowance for the years ended December 31, 2018 and 2017:
|
In thousands |
|
2018 |
|
|
2017 |
|
||
|
Beginning valuation allowance |
$ |
131,389 |
|
|
$ |
109,274 |
|
|
|
Increase as reflected in income tax expense |
|
13,609 |
|
|
|
11,466 |
|
|
|
Cumulative translation adjustment |
|
(4,881 |
) |
|
|
10,649 |
|
|
|
Ending valuation allowance |
$ |
140,117 |
|
|
$ |
131,389 |
|
|
During 2018, the Company recorded adjustments to its deferred tax accounts related to the impact of foreign exchange rate changes and to reconcile the financial statement accounts to the amounts expected to result in future income and deductions under local law, primarily as it relates to Irish net operating losses and deferred taxes for stock compensation. These adjustments were fully offset with valuation allowances based on the Company’s position with respect to the realizability of its recorded deferred tax assets in non-U.S. entities.
The Company has combined U.S., Irish, UK, and Israeli net operating loss carryforwards of $788.7 million, which do not expire. The total net operating loss carryforwards increased by approximately $78.3 million from the prior year primarily as a result of current year losses generated by the Company’s U.S. and Irish subsidiaries, the impact of foreign exchange rate changes, and adjustments to reconcile the financial statement accounts to the amounts reported on the filed 2017 foreign tax returns. In addition, the Company has U.S. Federal tax credit carryforwards of $7.1 million and state tax credit carryforwards of $2.1 million. These amounts exclude the impact of any unrecognized tax benefits and valuation allowances. These carryforwards, which will expire between 2024 and 2038, may be used to offset future taxable income.
As of December 31, 2018, earnings of $8.4 million have been retained indefinitely for reinvestment by foreign subsidiary or there is an expectation that any reinvestment can be recovered tax-free without significant cost, and the entity expects to ultimately use that means of recovery for domestic subsidiary companies; therefore, no provision has been made for income taxes that would be payable upon the distribution of such earnings and it would not be practicable to determine the amount of the related unrecognized deferred income tax liability.
The Company's and its subsidiaries' income tax returns are periodically examined by various taxing authorities. The Company is currently under audit by the New Jersey Department of Treasury for the years 2012 to 2015. Although the outcome of tax audits is always uncertain and could result in significant cash tax payments, the Company does not believe the outcome of these audits will have a material adverse effect on the Company's consolidated financial position or results of operations.
(11) Stock Incentive Plans and Stock-Based Compensation
On April 29, 2011 the Board, upon the recommendation of the Remuneration Committee, adopted the 2011 Stock Incentive Plan (“2011 Plan”), which was approved by the Company’s shareholders on July 12, 2011. The 2011 Plan replaced the Company’s 2002 Stock Option Plan (“2002 Plan”), which expired on January 1, 2012. The maximum number of the Company’s Ordinary Shares of £0.50 each or any ADS’s, as to be issued under the 2011 Plan, as amended, shall not exceed the sum of (i) 51.5 million newly authorized Shares available for award and (ii) the number of Shares that remained available for grants under the Company’s 2002 Plan and (iii) the number of Shares underlying then outstanding awards under the 2002 Plan that could be subsequently forfeited, cancelled, expire or are otherwise terminated. The award of stock options (both incentive and non-qualified options) and restricted stock units, and awards of unrestricted Shares to Directors are permitted. The 2011 Plan is administered by the Remuneration Committee of the Company’s Board of Directors and expires on July 12, 2021.
In addition to the grants under the 2011 Plan, the Company grants non-qualified stock options to employees to purchase the Company’s ordinary shares. These grants are made pursuant to employment agreements on terms consistent with the 2011 Plan.
Under the terms of the 2011 Plan, and grants made pursuant to employment agreements, options typically vest over a four-year period, expire after a ten-year term and are granted at an exercise price equal to the closing price of the Company’s American Depositary Shares on the grant date. The following table summarizes all stock option activity for the year ended December 31, 2018:
F-28
|
In thousands (except per share amounts and years) |
Number of Shares |
|
|
Weighted Average Exercise Price |
|
|
Weighted Average Remaining Contractual Term |
|
Aggregate Intrinsic Value |
|
|||
|
Outstanding as of January 1, 2018 |
|
24,108 |
|
|
$ |
3.26 |
|
|
|
|
|
|
|
|
Granted |
|
4,363 |
|
|
|
7.82 |
|
|
|
|
|
|
|
|
Forfeited |
|
(1,058 |
) |
|
|
3.39 |
|
|
|
|
|
|
|
|
Expired |
|
(12 |
) |
|
|
3.40 |
|
|
|
|
|
|
|
|
Exercised |
|
(8,138 |
) |
|
|
3.24 |
|
|
|
|
|
|
|
|
Outstanding as of December 31, 2018 |
|
19,263 |
|
|
|
4.29 |
|
|
6.5 years |
|
$ |
188,022 |
|
|
Exercisable as of December 31, 2018 |
|
12,415 |
|
|
|
3.53 |
|
|
5.4 years |
|
|
125,395 |
|
|
Vested and expected to vest as of December 31, 2018 |
|
18,921 |
|
|
$ |
4.27 |
|
|
6.5 years |
|
$ |
184,900 |
|
|
Available for future grant as of December 31, 2018 |
|
11,463 |
|
|
|
|
|
|
|
|
|
|
|
The weighted average grant date fair value of stock options granted during the years ended December 31, 2018, 2017 and 2016 was $7.82, $3.09, and $1.62, respectively. The total grant date fair value of options vested during the years ended December 31, 2018, 2017 and 2016 was $7.7 million, $7.1 million, and $6.5 million, respectively.
During the years ended December 31, 2018, 2017 and 2016, the Company received proceeds from the exercise of options of $26.4 million, $0.6 million, and $0.3 million, respectively. The total intrinsic value of options exercised during the years ended December 31, 2018, 2017 and 2016 was $69.7 million, $0.5 million, and $0.2 million, respectively, calculated as the difference between the quoted stock price of the Company’s common stock as of the reporting date and the exercise prices of the underlying awards.
As of December 31, 2018, there was $25.7 million of unrecognized stock-based compensation expense related to unvested stock option share-based compensation arrangements granted under the Company’s stock award plans. This expense is expected to be recognized over a weighted-average period of approximately 2.3 years. The Company recognizes compensation expense for the fair values of those awards which have graded vesting on a straight-line basis.
The fair value of stock options on the date of grant was estimated using the Black-Scholes option pricing model. Use of a valuation model requires management to make certain assumptions with respect to selected model inputs. Expected stock price volatility was calculated based on the historical volatility of the Company’s common stock over the expected life of the option. The expected life was determined using the simplified method based on the term and vesting period. The risk-free interest rate is based on zero-coupon U.S. Treasury securities with a maturity term approximating the expected life of the option at the date of grant. No dividend yield has been assumed as the Company does not currently pay dividends on its common stock and does not anticipate doing so in the foreseeable future. Estimated forfeitures are based on the Company’s historical forfeiture activity.
Employee stock options generally vest over a four-year service period and all stock options are settled by the issuance of new shares. Compensation expense recognized for all option grants is net of estimated forfeitures and is recognized over the awards’ respective requisite service periods. The vesting of certain stock options is contingent upon the attainment of performance criteria. The probability that such criteria will be achieved is assessed by management and compensation expense for such awards is only recorded to the extent that the attainment of the performance criteria is deemed to be probable. The Company recorded compensation expense in relation to stock options of $8.2 million, $7.0 million and $6.6 million for the years ended December 31, 2018, 2017 and 2016, respectively.
For 2018, 2017 and 2016, the Company used the following assumptions to estimate the fair value of share-based payment awards:
|
|
2018 |
|
|
2017 |
|
|
2016 |
|
|
Risk-free interest rate |
2.18% - 3.00% |
|
|
1.77% - 2.01% |
|
|
1.07% - 1.70% |
|
|
Expected dividend yield |
0.00% |
|
|
0.00% |
|
|
0.00% |
|
|
Expected option life (years) |
6.25 |
|
|
6.25 |
|
|
6.25 |
|
|
Expected volatility |
71% - 92% |
|
|
73% - 82% |
|
|
83% - 86% |
|
F-29
Restricted Stock Units
The 2011 Plan also allows for granting of restricted stock unit awards under the terms of the Plan. The restricted stock units vest based upon a time-based service condition, a performance condition, or both. The probability that any performance criteria will be achieved is assessed by management and compensation expense for such awards is only recorded to the extent that the attainment of the performance criteria is deemed to be probable. Restricted stock units are recorded as compensation expense based on fair value, representing the market value of the Company’s common stock on the date of grant. The fair value of restricted stock units is amortized on a straight-line basis through the statement of operations over the service period until the shares have vested. The following table presents the restricted stock unit activity for the years ended December 31, 2018 and 2017:
|
In thousands (except per share amounts) |
Shares |
|
|
Weighted Average Grant Date Fair Value |
|
||
|
Outstanding as of January 1, 2017 |
|
10,143 |
|
|
$ |
2.09 |
|
|
Granted |
|
4,197 |
|
|
|
3.04 |
|
|
Vested |
|
(2,179 |
) |
|
|
1.62 |
|
|
Forfeited |
|
(155 |
) |
|
|
2.91 |
|
|
Outstanding as of December 31, 2017 |
|
12,006 |
|
|
|
2.50 |
|
|
Granted |
|
2,633 |
|
|
|
4.32 |
|
|
Vested |
|
(4,611 |
) |
|
|
2.20 |
|
|
Forfeited |
|
(395 |
) |
|
|
3.43 |
|
|
Outstanding as of December 31, 2018 |
|
9,633 |
|
|
$ |
3.12 |
|
The Company recorded compensation expense in relation to restricted stock units of $10.6 million, $7.0 million and $7.0 million for the years ended December 31, 2018, 2017 and 2016 respectively.
The following table presents the stock-based compensation expense related to stock-based awards for the years ended December 31, 2018, 2017 and 2016:
|
In thousands |
2018 |
|
|
2017 |
|
|
2016 |
|
|||
|
Research and development |
$ |
2,898 |
|
|
$ |
2,122 |
|
|
$ |
2,252 |
|
|
Selling, general and administrative |
|
15,908 |
|
|
|
11,838 |
|
|
|
11,361 |
|
|
Stock-based compensation expense |
$ |
18,806 |
|
|
$ |
13,960 |
|
|
$ |
13,613 |
|
Employee Stock Purchase Plan
On March 13, 2017, the Board adopted, subject to shareholder approval, the Amarin Corporation plc 2017 Employee Stock Purchase Plan (the “ESPP”), which was approved by the Company’s shareholders on May 15, 2017. The ESPP is intended to qualify as an “employee stock purchase plan” within the meaning of Section 423 of the Internal Revenue Code. The maximum fair market value of stock which can be purchased by a participant in a calendar year is $25,000. Under the ESPP, an aggregate of 3,000,000 ordinary shares (each ordinary share to be represented by one ADS) are reserved and available for issuance, which were registered with the SEC on August 2, 2017, for sale to eligible employees. Subject to certain exclusions, any employee of the Company’s U.S. subsidiary, Amarin Pharma, Inc., who works at least 20 hours per week and has been employed for at least six months as of the first day of the applicable offering period is eligible to participate in the ESPP. Eligible employees may authorize payroll deductions of up to 15 percent of their base pay to be withheld to purchase ordinary shares, subject to terms and limitations of the plan, at a price equal to 85 percent of the lower of the fair market values of the Company’s ordinary shares as of the beginning or the end of six-month offering periods. For the offering periods ended May 31, 2018 and November 30, 2018, the Company issued 127,872 shares and 184,385 shares, respectively, at a purchase price of $2.81 per share and $2.86 per share, respectively. As of December 31, 2018, 2,687,743 shares were reserved for future issuance under the ESPP. No shares were issued under the ESPP during the year ended December 31, 2017 as the initial enrollment and offering periods commenced in 2018.
(12) Defined Contribution Plan
The Company makes available a 401(k) plan for its U.S. employees. Under the 401(k) plan, employees may make contributions which are eligible for a discretionary percentage match, in cash, as defined in the 401(k) plan and determined by the Board of Directors. The Company recognized $0.7 million, $0.4 million and $0.5 million of related compensation expense for the year ended December 31, 2018, 2017 and 2016, respectively.
F-30
(13) Quarterly Summarized Financial Information (Unaudited)
|
|
Fiscal years ended December 31, 2018 and 2017 |
|
||||||||||||||||||||||||||||||
|
|
1st Quarter |
|
|
2nd Quarter |
|
|
3rd Quarter |
|
|
4th Quarter |
|
|||||||||||||||||||||
|
|
2018 |
|
|
2017 |
|
|
|
2018 |
|
|
2017 |
|
|
2018 |
|
|
2017 |
|
|
2018 |
|
|
2017 |
|
||||||||
|
|
(In thousands, except per share amounts) |
|
||||||||||||||||||||||||||||||
|
Total revenue, net |
$ |
43,919 |
|
|
$ |
34,637 |
|
|
|
$ |
52,643 |
|
|
$ |
45,241 |
|
|
$ |
55,323 |
|
|
$ |
47,360 |
|
|
$ |
77,330 |
|
|
$ |
53,866 |
|
|
Net loss |
|
(24,095 |
) |
|
|
(20,941 |
) |
|
|
|
(34,210 |
) |
|
|
(13,634 |
) |
|
|
(24,471 |
) |
|
|
(10,825 |
) |
|
|
(33,670 |
) |
|
|
(22,465 |
) |
|
Loss per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
$ |
(0.08 |
) |
|
$ |
(0.08 |
) |
|
|
$ |
(0.12 |
) |
|
$ |
(0.05 |
) |
|
$ |
(0.08 |
) |
|
$ |
(0.04 |
) |
|
$ |
(0.11 |
) |
|
$ |
(0.08 |
) |
|
Diluted |
$ |
(0.08 |
) |
|
$ |
(0.08 |
) |
|
|
$ |
(0.12 |
) |
|
$ |
(0.05 |
) |
|
$ |
(0.08 |
) |
|
$ |
(0.04 |
) |
|
$ |
(0.11 |
) |
|
$ |
(0.08 |
) |
On March 31, 2014, the Company entered into a Co-Promotion Agreement (the “Agreement”) with Kowa Pharmaceuticals America, Inc. related to the commercialization of Vascepa® (icosapent ethyl) capsules in the United States. Under the terms of the Agreement, Amarin granted to Kowa Pharmaceuticals America, Inc. the right to be the sole co-promoter, together with the Company, of Vascepa in the United States during the term. The Agreement was amended on July 25, 2017 to reflect evolving promotional needs, including refinement of target lists. Amarin and Kowa Pharmaceuticals America, Inc. intentionally designed the co-promotion to naturally end as of December 31, 2018 and mutually agreed not to renew the agreement.
During the term, Kowa Pharmaceuticals America, Inc. and Amarin agreed to use commercially reasonable efforts to promote, detail and optimize sales of Vascepa in the United States. The performance requirements included a negotiated minimum number of details to be delivered by each party in the first and second position, and the use of a negotiated number of minimum sales representatives from each party. Kowa Pharmaceuticals America, Inc. agreed to bear the costs incurred for its sales force associated with the commercialization of Vascepa and to pay for certain incremental costs associated with the use of its sales force, such as sample costs and costs for promotional and marketing materials. Amarin recognized all revenue from sales of Vascepa and used commercially reasonable efforts to maintain a minimum amount of inventory of Vascepa for use in the United States.
In exchange for Kowa Pharmaceuticals America, Inc.’s co-promotional services, Kowa Pharmaceuticals America, Inc. was entitled to a quarterly co-promotion fee based on aggregate Vascepa gross margin that varied during the term. The percentage of aggregate Vascepa gross margin earned by Kowa Pharmaceuticals America, Inc. was, as amended, approximately eighteen percent (18%) in 2017, partially offset by certain other refinements. During 2018, which was the last year of the Agreement, as amended, the Company incurred expense for both the annual co-promotion fee, which in 2018 was calculated as eighteen-and-a-half percent (18.5%) of Vascepa gross margin, plus accrual for co-promotion tail payments which were calculated as a percentage of the 2018 co-promotion fee. Kowa Pharmaceuticals America, Inc. is eligible to receive $17.8 million in co-promotion tail payments, the present value of which of $16.6 million was fully accrued as of December 31, 2018. No amount was accrued for the co-promotion tail payments as of December 31, 2017. The accrued tail payments will be paid over three years with declining amounts each year.
As of December 31, 2018 and 2017, the Company had a net payable to Kowa Pharmaceuticals America, Inc. of $27.6 million and $8.3 million, respectively, of which $18.1 million and $8.3 million, respectively, was classified as current on the consolidated balance sheets, representing co-promotion fees, including accrual of the tail payments, net of reimbursable amounts incurred for samples and other marketing expenses.
(15) Revenue Recognition
The Company sells Vascepa principally to a limited number of major wholesalers, as well as selected regional wholesalers and specialty pharmacy providers in the United States, or collectively, its Distributors or its Customers, that in turn resell Vascepa to retail pharmacies for subsequent resale to patients and healthcare providers. Patients are required to have a prescription in order to purchase Vascepa. In addition to distribution agreements with Distributors, the Company enters into arrangements with health care providers and payors that provide for government-mandated and/or privately-negotiated rebates, chargebacks and discounts with respect to the purchase of the Company’s product.
Revenues from product sales are recognized when the Distributor obtains control of the Company’s product, which occurs at a point in time, typically upon delivery to the Distributor. Payments from Distributors are generally received 30-60 days from date of sale. The Company evaluates the creditworthiness of each of its Distributors to determine whether revenues can be recognized upon delivery, subject to satisfaction of the other requirements, or whether recognition is required to be delayed until receipt of payment. The Company calculates gross product revenues generally based on the wholesale acquisition cost that the Company charges its Distributors for Vascepa.
F-31
Reserves for Variable Consideration
Revenues from product sales are recorded at the net sales price (transaction price), which includes estimates of variable consideration for which reserves are established and which result from (a) trade allowances, such as invoice discounts for prompt pay and distributor fees, (b) estimated government and private payor rebates and chargebacks and discounts, such as Medicaid reimbursements, (c) reserves for expected product returns and (d) estimated costs of incentives that are offered within contracts between the Company and its Distributors, health care providers, payors and other indirect customers relating to the Company’s sales of its product. These reserves are based on the amounts earned or to be claimed on the related sales and are classified as reductions of accounts receivable (if the amount is payable to the Distributor) or as a current liability (if the amount is payable to a party other than a Distributor). Where appropriate, these estimates take into consideration a range of possible outcomes which are probability-weighted for relevant factors such as the Company’s historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. Overall, these reserves reflect the Company’s best estimates of the amount of consideration to which it is entitled based on the terms of the contract. The amount of variable consideration which is included in the transaction price may be constrained, and is included in the net sales price only to the extent that it is probable that a significant reversal in the amount of the cumulative revenue recognized will not occur in a future period. Actual amounts of consideration ultimately received may differ from the Company’s estimates. If actual results in the future vary from the Company’s estimates, the Company adjusts these estimates, which would affect net product revenue and earnings in the period such variances become known.
Trade Allowances: The Company generally provides invoice discounts on Vascepa sales to its Distributors for prompt payment and fees for distribution services, such as fees for certain data that Distributors provide to the Company. The payment terms for sales to Distributors generally include a 2% discount for prompt payment while the fees for distribution services are based on contractual rates agreed with the respective Distributors. Based on historical data, the Company expects its Distributors to earn these discounts and fees, and deducts the full amount of these discounts and fees from its gross product revenues and accounts receivable at the time such revenues are recognized.
Rebates, Chargebacks and Discounts: The Company contracts with Medicaid, Medicare, other government agencies and various private organizations, or collectively, Third-party Payors, so that Vascepa will be eligible for purchase by, or partial or full reimbursement from, such Third-party Payors. The Company estimates the rebates, chargebacks and discounts it will provide to Third-party Payors and deducts these estimated amounts from its gross product revenues at the time the revenues are recognized. The Company estimates these reserves based upon a range of possible outcomes that are probability-weighted for the estimated payor mix. These reserves are recorded in the same period the revenue is recognized, resulting in a reduction of product revenue and the establishment of a current liability, which is included in accrued expenses and other current liabilities on the consolidated balance sheets. For Medicare, the Company also estimates the number of patients in the prescription drug coverage gap for whom the Company will owe an additional liability under the Medicare Part D program. The Company estimates the rebates, chargebacks and discounts that it will provide to Third-party Payors based upon (i) the Company’s contracts with these Third-party Payors, (ii) the government-mandated discounts applicable to government-funded programs, (iii) information obtained from the Company’s Distributors and (iv) information obtained from other third parties regarding the payor mix for Vascepa. The Company’s liability for these rebates consists of invoices received for claims from prior quarters that have not been paid or for which an invoice has not yet been received, estimates of claims for the current quarter, and estimated future claims that will be made for product that has been recognized as revenue, but remains in the distribution channel inventories at the end of each reporting period.
Product Returns: The Company’s Distributors have the right to return unopened unprescribed Vascepa during the 18-month period beginning six months prior to the labeled expiration date and ending twelve months after the labeled expiration date. The expiration date for Vascepa 1-gram and 0.5-gram size capsules is currently four years and three years, respectively, after being converted into capsule form, which is the last step in the manufacturing process for Vascepa and generally occurs within a few months before Vascepa is delivered to Distributors. The Company estimates future product returns on sales of Vascepa based on: (i) data provided to the Company by its Distributors (including weekly reporting of Distributors’ sales and inventory held by Distributors that provided the Company with visibility into the distribution channel in order to determine what quantities were sold to retail pharmacies and other providers), (ii) information provided to the Company from retail pharmacies, (iii) data provided to the Company by a third-party data provider which collects and publishes prescription data, and other third parties, (iv) historical industry information regarding return rates for similar pharmaceutical products, (v) the estimated remaining shelf life of Vascepa previously shipped and currently being shipped to Distributors and (vi) contractual agreements intended to limit the amount of inventory maintained by the Company’s Distributors. These reserves are recorded in the same period the related revenue is recognized, resulting in a reduction of product revenue and the establishment of a current liability which is included in accrued expenses and other current liabilities on the consolidated balance sheets.
F-32
Other Incentives: Other incentives that the Company offers to indirect customers include co-pay mitigation rebates provided by the Company to commercially insured patients who have coverage for Vascepa and who reside in states that permit co-pay mitigation programs. The Company’s co-pay mitigation program is intended to reduce each participating patient’s portion of the financial responsibility for Vascepa’s purchase price to a specified dollar amount. Based upon the terms of the program and information regarding programs provided for similar specialty pharmaceutical products, the Company estimates the average co-pay mitigation amounts and the percentage of patients that it expects to participate in the program in order to establish its accruals for co-pay mitigation rebates. These reserves are recorded in the same period the related revenue is recognized, resulting in a reduction of product revenue and the establishment of a current liability which is included in accrued expenses and other current liabilities on the consolidated balance sheets. The Company adjusts its accruals for co-pay mitigation rebates based on actual redemption activity and estimates regarding the portion of issued co-pay mitigation rebates that it estimates will be redeemed.
The following table summarizes activity in each of the net product revenue allowance and reserve categories described above for the years ended December 31, 2018 and 2017:
|
In thousands |
|
Trade Allowances |
|
|
Rebates, Chargebacks and Discounts |
|
|
Product Returns |
|
|
Other Incentives |
|
|
Total |
|
|||||
|
Balance as of January 1, 2017 |
|
$ |
3,743 |
|
|
$ |
20,915 |
|
|
$ |
859 |
|
|
$ |
1,681 |
|
|
$ |
27,198 |
|
|
Provision related to current period sales |
|
|
35,067 |
|
|
|
126,903 |
|
|
|
1,480 |
|
|
|
15,081 |
|
|
|
178,531 |
|
|
Provision related to prior period sales |
|
|
(323 |
) |
|
|
(682 |
) |
|
|
— |
|
|
|
(70 |
) |
|
|
(1,075 |
) |
|
Credits/payments made for current period sales |
|
|
(23,087 |
) |
|
|
(96,181 |
) |
|
|
(344 |
) |
|
|
(12,773 |
) |
|
|
(132,385 |
) |
|
Credits/payments made for prior period sales |
|
|
(3,365 |
) |
|
|
(18,891 |
) |
|
|
(108 |
) |
|
|
(1,812 |
) |
|
|
(24,176 |
) |
|
Balance as of December 31, 2017 |
|
|
12,035 |
|
|
|
32,064 |
|
|
|
1,887 |
|
|
|
2,107 |
|
|
|
48,093 |
|
|
Provision related to current period sales |
|
|
46,002 |
|
|
|
190,329 |
|
|
|
1,211 |
|
|
|
20,732 |
|
|
|
258,274 |
|
|
Provision related to prior period sales |
|
|
— |
|
|
|
(1,845 |
) |
|
|
— |
|
|
|
(69 |
) |
|
|
(1,914 |
) |
|
Credits/payments made for current period sales |
|
|
(29,202 |
) |
|
|
(148,857 |
) |
|
|
— |
|
|
|
(19,307 |
) |
|
|
(197,366 |
) |
|
Credits/payments made for prior period sales |
|
|
(9,340 |
) |
|
|
(30,057 |
) |
|
|
(150 |
) |
|
|
(2,296 |
) |
|
|
(41,843 |
) |
|
Balance as of December 31, 2018 |
|
$ |
19,495 |
|
|
$ |
41,634 |
|
|
$ |
2,948 |
|
|
$ |
1,167 |
|
|
$ |
65,244 |
|
Such net product revenue allowances and reserves are included within accrued expenses and other current liabilities within the consolidated balance sheets, with the exception of trade allowances and chargebacks, which are included within accounts receivable, net as discussed below.
Licensing Revenue
The Company enters into licensing agreements which are within the scope of Topic 606, Revenue from Contracts with Customers, under which it licenses certain rights to Vascepa for uses that are currently commercialized and under development by the Company. The terms of these arrangements typically include payment to the Company of one or more of the following: non-refundable, up-front license fees; development, regulatory and commercial milestone payments; payments for manufacturing supply services the Company provides through its contract manufacturers; and royalties on net sales of licensed products. Each of these payments results in licensing revenues.
In determining the appropriate amount of revenue to be recognized as it fulfills its obligations under each of its agreements, the Company performs the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) the Company satisfies each performance obligation.
In determining performance obligations, management evaluates whether the license is distinct from the other performance obligations with the collaborative partner based on the consideration of the relevant facts and circumstances for each arrangement. Factors considered in the determination of include the stage of development of the license delivered, research and development capabilities of the partner and the ability of partners to develop and commercialize Vascepa independent of the Company.
Licenses of intellectual property: If the license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, the Company recognizes revenues from non-refundable, up-front fees allocated to the license when the license is transferred to the customer and the customer is able to use and benefit from the license. For licenses that are bundled with other promises, the Company utilizes judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the appropriate
F-33
method of measuring progress for purposes of recognizing revenue from non-refundable, up-front fees. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the measure of performance and related revenue recognition.
Milestone Payments: At the inception of each arrangement that includes development, regulatory and commercial milestone payments, the Company evaluates whether the milestones are considered probable of being reached and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within the control of the Company or licensee, such as regulatory approvals, are not considered probable of being achieved until those approvals are received. The Company evaluates factors such as the scientific, clinical, regulatory, commercial and other risks that must be overcome to achieve the respective milestone as well as the level of effort and investment required. The transaction price is then allocated to each performance obligation on a relative stand-alone selling price basis, for which the Company recognizes revenue as or when the performance obligations under the contract are satisfied. At the end of each subsequent reporting period, the Company re-evaluates the probability of achievement of such development, regulatory and commercial milestones and any related constraint, and if necessary, adjusts its estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect licensing revenues and earnings in the period of adjustment.
The Company receives payments from its customers based on billing schedules established in each contract. Up-front payments and fees are recorded as deferred revenue upon receipt or when due, and may require deferral of revenue recognition to a future period until the Company performs its obligations under these arrangements. Amounts are recorded as accounts receivable when the Company’s right to consideration is unconditional. The Company does not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the customer and the transfer of the promised goods or services to the customer will be one year or less.
(16) Development, Commercialization and Supply Agreements
In-licenses
Mochida Pharmaceutical Co., Ltd.
In June 2018, the Company entered into a collaboration with Mochida Pharmaceutical Co., Ltd. (“Mochida”) related to the development and commercialization of drug products and indications based on the active pharmaceutical ingredient in Vascepa, the omega-3 acid, EPA (eicosapentaenoic acid). Among other terms in the agreement, the Company obtained an exclusive license to certain Mochida intellectual property to advance the Company’s interests in the United States and certain other territories and the parties will collaborate to research and develop new products and indications based on EPA for the Company’s commercialization in the United States and certain other territories. The potential new product and indication opportunities contemplated under this agreement are currently in early stages of development.
Upon closing of the collaboration agreement, the Company made a non-refundable, non-creditable upfront payment of approximately $2.7 million, which was recorded in research and development expense in the consolidated statement of operations for the year ended December 31, 2018. In addition, the agreement provides for the Company to pay milestone payments upon the achievement of certain product development milestones and royalties on net sales of future products arising from the collaboration, if any.
Out-licenses
Eddingpharm (Asia) Macao Commercial Offshore Limited
In February 2015, the Company entered into a Development, Commercialization and Supply Agreement (the “DCS Agreement”) with Eddingpharm (Asia) Macao Commercial Offshore Limited (“Eddingpharm”) related to the development and commercialization of Vascepa in Mainland China, Hong Kong, Macau and Taiwan, or the “China Territory.” Under the terms of the DCS Agreement, the Company granted to Eddingpharm an exclusive (including as to the Company) license with right to sublicense to develop and commercialize Vascepa in the China Territory for uses that are currently commercialized and under development by the Company based on the Company’s MARINE, ANCHOR and REDUCE-IT clinical trials of Vascepa.
Under the DCS Agreement, Eddingpharm is solely responsible for development and commercialization activities in the China Territory and associated expenses. The Company provides development assistance and is responsible for supplying finished and later bulk drug product at defined prices under negotiated terms. The Company retains all Vascepa manufacturing rights. Eddingpharm agreed to certain restrictions regarding the commercialization of competitive products globally and the Company agreed to certain restrictions regarding the commercialization of competitive products in the China Territory.
The Company and Eddingpharm agreed to form a joint development committee to oversee regulatory and development activities for Vascepa in the China Territory in accordance with a negotiated development plan and to form a separate joint commercialization committee to oversee Vascepa commercialization activities in the China Territory. Development costs are paid by Eddingpharm to the
F-34
extent such costs are incurred in connection with the negotiated development plan or otherwise incurred by Eddingpharm. Eddingpharm is responsible for preparing and filing regulatory applications in all countries of the China Territory at Eddingpharm’s cost with the Company’s assistance. The DCS Agreement also contains customary provisions regarding indemnification, supply, record keeping, audit rights, reporting obligations, and representations and warranties that are customary for an arrangement of this type.
The term of the DCS Agreement expires, on a product-by-product basis, upon the later of (i) the date on which such product is no longer covered by a valid claim under a licensed patent in the China Territory, or (ii) the twelfth (12th) anniversary of the first commercial sale of such product in Mainland China. The DCS Agreement may be terminated by either party in the event of a bankruptcy of the other party and for material breach, subject to customary cure periods. In addition, at any time following the third anniversary of the first commercial sale of a product in Mainland China, Eddingpharm has the right to terminate the DCS Agreement for convenience with twelve months’ prior notice. Neither party may assign or transfer the DCS Agreement without the prior consent of the other party, provided that the Company may assign the DCS Agreement in the event of a change of control transaction.
Upon closing of the DCS Agreement, the Company received a non-refundable $15.0 million up-front payment. In March 2016, Eddingpharm submitted its clinical trial application (“CTA”) with respect to the MARINE indication for Vascepa to the Chinese regulatory authority. Following the CTA submission, the Company received a non-refundable $1.0 million milestone payment. In March 2017, the CTA was approved by the Chinese regulatory authority, and, in December 2017, Eddingpharm commenced a pivotal clinical trial aimed to support the regulatory approval of the first indication of Vascepa in a patient population with severe hypertriglyceridemia in Mainland China.
In addition to the non-refundable, up-front and regulatory milestone payments described above, the Company is entitled to receive certain regulatory and sales-based milestone payments of up to an additional $153.0 million as well as tiered double-digit percentage royalties on net sales of Vascepa in the China Territory escalating to the high teens. The regulatory milestone events relate to the submission and approval of certain applications to the applicable regulatory authority, such as a clinical trial application, clinical trial exemption, or import drug license application. The amounts to be received upon achievement of the regulatory milestone events relate to the submission and approval for three indications, and range from $2.0 million to $15.0 million for a total of $33.0 million. The sales-based milestone events occur when annual aggregate net sales of Vascepa in the territory equals or exceeds certain specified thresholds, and range from $5.0 million to $50.0 million for a total of $120.0 million. Each such milestone payment shall be payable only once regardless of how many times the sales milestone event is achieved. Each such milestone payment is non-refundable and non-creditable against any other milestone payments.
The Company assessed this arrangement in accordance with Topic 606 and concluded that the contract counterparty, Eddingpharm, is a customer. The Company identified the following performance obligations at the inception of the DCS Agreement: (1) the exclusive license to develop and commercialize Vascepa in the China Territory for uses that are currently commercialized and under development by the Company, (2) the obligation to participate in various steering committees, (3) ongoing development and regulatory assistance, and (4) manufacture and supply of commercial product. Based on the analysis performed, the Company concluded that the identified performance obligations are not distinct and therefore a combined performance obligation.
The transaction price includes the $15.0 million up-front consideration received and the $1.0 million milestone payment received related to the successful submission of the CTA for the MARINE indication. None of the other clinical or regulatory milestones have been included in the transaction price, as all milestone amounts were fully constrained. As part of its evaluation of the constraint, the Company considered numerous factors, including that receipt of the milestones is outside the control of the Company and contingent upon success in future clinical trials and the licensee’s efforts. Any consideration related to sales-based milestones (including royalties) will be recognized when the related sales occur as they were determined to relate predominantly to the license granted to Eddingpharm and therefore have also been excluded from the transaction price. The Company will re-evaluate the transaction price in each reporting period and as uncertain events are resolved or other changes in circumstances occur.
During the years ended December 31, 2018 and 2017, the Company recognized $0.1 million and $1.2 million, respectively, as licensing revenue related to the up-front and milestone payments received in connection with the Eddingpharm agreement. Through December 31, 2018 and 2017, the Company has recognized $2.8 million and $3.1 million, respectively, as licensing revenue under the DCS Agreement concurrent with the support provided by Amarin to Eddingpharm in achieving the combined performance obligation, which in the Company’s judgment is the best measure of progress towards satisfying the performance obligation. The remaining transaction price of $13.2 million and $12.9 million is recorded in deferred revenue as of December 31, 2018 and 2017, respectively, on the consolidated balance sheets and will be recognized as revenue over the remaining period of 16 years.
Biologix FZCo
In March 2016, the Company entered into an agreement with Biologix FZCo (“Biologix”), a company incorporated under the laws of the United Arab Emirates, to register and commercialize Vascepa in several Middle Eastern and North African countries. Under the
F-35
terms of the distribution agreement, the Company granted to Biologix a non-exclusive license to use its trademarks in connection with the importation, distribution, promotion, marketing and sale of Vascepa in the Middle East and North Africa territory. Upon closing of the agreement, the Company received a non-refundable up-front payment, which will be recognized as revenue over 10 years commencing upon first marketing approval of Vascepa in the territory. The Company is entitled to receive all payments based on total product sales and pays Biologix a service fee in exchange for its services, whereby the service fee represents a percentage of gross selling price which is subject to a minimum floor price.
In March 2018 and July 2018, the Company received approval for Vascepa as a prescription medication for use in Lebanon and United Arab Emirates, respectively, as an adjunct to diet to reduce triglyceride levels in adult patients with severe hypertriglyceridemia.
HLS Therapeutics, Inc.
In September 2017, the Company entered into an agreement with HLS Therapeutics Inc. (“HLS”), a company incorporated under the laws of Canada, to register, commercialize and distribute Vascepa in Canada. Under the agreement, HLS will be responsible for regulatory and commercialization activities and associated costs. The Company is responsible for providing assistance towards local filings, supplying finished product under negotiated supply terms, maintaining intellectual property, and continuing the development and funding of REDUCE-IT related activities.
Upon closing of the agreement, the Company received one-half of a non-refundable $5.0 million up-front payment, and received the remaining half on the six-month anniversary of the closing. Following achievement of the REDUCE-IT trial primary endpoint, which was announced in September 2018, the Company is entitled to receive a non-refundable $2.5 million milestone payment which is recorded in other receivables on the consolidated balance sheet as of September 30, 2018. In addition to the non-refundable, up-front and regulatory milestone payments just described, the Company is entitled to receive certain regulatory and sales-based milestone payments of up to an additional $57.5 million, as well as tiered double-digit royalties on net sales of Vascepa in Canada.
The Company assessed this arrangement in accordance with Topic 606 and concluded that the contract counterparty, HLS, is a customer. The Company identified the following performance obligations at the inception of the contract: (1) license to HLS to develop, register, and commercialize Vascepa in Canada, (2) support general development and regulatory activities, (3) participate in various steering committees, and (4) manufacture and provide finished form of product. Based on the analysis performed, the Company concluded that the identified performance obligations in the agreement are not distinct and therefore a combined performance obligation.
The transaction price includes the $5.0 million up-front consideration and the $2.5 million milestone related to the achievement of the REDUCE-IT trial primary endpoint. None of the other regulatory milestones have been included in the transaction price, as all of the remaining regulatory milestone amounts were fully constrained. As part of its evaluation of the constraint, the Company considered numerous factors, including that receipt of the milestones is outside the control of the Company and contingent upon the licensee’s efforts. Any consideration related to sales-based milestones (including royalties) will be recognized when the related sales occur as they were determined to relate predominantly to the license granted to HLS and therefore have also been excluded from the transaction price. The Company will re-evaluate the transaction price in each reporting period and as uncertain events are resolved or other changes in circumstances occur.
During the years ended December 31, 2018 and 2017, the Company recognized $0.6 million and $0.1 million, respectively, as licensing revenue related to up-front and milestone payments received in connection with the HLS agreement. Through December 31, 2018 and 2017, the Company has recognized $0.9 million and $0.1 million, respectively, as licensing revenue under the agreement concurrent with the support provided by Amarin to HLS in achieving the performance obligation, which in the Company’s judgment is the best measure of progress towards satisfying the combined performance obligation. The remaining transaction price of $6.6 million and $4.9 million is recorded in deferred revenue as of December 31, 2018 and 2017, respectively, on the consolidated balance sheets and will be recognized as revenue over the remaining period of 12 years.
The following table presents changes in the balances of the Company’s contract assets and liabilities during the nine months ended December 31, 2018:
|
In thousands |
|
Balance at Beginning of Period |
|
|
Additions |
|
|
Deductions |
|
Balance at End of Period |
|
Year ended December 31, 2018: |
|
|
|
|
|
|
|
|
|
|
|
Contract assets |
$ |
— |
|
$ |
— |
|
$ |
— |
$ |
— |
|
Contract liabilities: |
|
|
|
|
|
|
|
|
|
|
|
Deferred revenue |
$ |
19,054 |
|
$ |
2,500 |
|
$ |
(843) |
$ |
20,710 |
F-36
During the year ended December 31, 2018, the Company recognized the following revenues as a result of changes in the contract asset and contract liability balances in the respective periods:
|
In thousands |
|
Year Ended December 31, |
|
|
Revenue recognized in the period from: |
|
2018 |
|
|
Amounts included in contract liability at the beginning of the period |
|
$ |
650 |
|
Performance obligations satisfied in previous periods |
|
$ |
193 |
(17) Subsequent Events
On February 22, 2019, a class action lawsuit was filed against Amarin Corporation plc, the Company’s chief executive officer and chief scientific officer in the U.S. District Court for the District of New Jersey. See Note 8—Commitments and Contingencies for further information.
On February 5, 2019, the Company entered into a lease agreement for approximately 67,747 square feet of new office space in Bridgewater, New Jersey (the “Lease”). The Lease will commence upon delivery of the premises after certain improvements are made, which is anticipated to be on or about July 1, 2019 (the “Commencement Date”) for an 11-year period, with two five-year renewal options. Subject to the terms of the Lease, Amarin will have a one-time option to terminate the agreement effective on the first day of the ninety-seventh month after the Commencement Date upon advanced written notice and a termination payment specified in the Lease. Under the Lease, the Company will pay monthly rent of approximately $141,000 for the first year following the Commencement Date, and such rent will increase by a nominal percentage every year following the first anniversary of the Commencement Date.
F-37
EXHIBIT 10.69
LEASE
between
440 Route 22 LLC
as Landlord
and
AMARIN PHARMA, INC.
as Tenant
For Premises Located at
440 US Highway 22
Bridgewater, New Jersey
1
THIS LEASE (this “Lease”) is made and dated as of the 5th day of February, 2019 by and between 440 Route 22 LLC, a New Jersey limited liability company, having an address at c/o Broad Real Estate Services, 910 East County Line Road, Suite 202A, Lakewood, New Jersey 08701 (“Landlord”), and Amarin Pharma, Inc., a Delaware corporation, having an address at 1430 Route 206, Bedminster, New Jersey 07921 (“Tenant”).
W I T N E S S E T H:
ARTICLE 1
BASIC DATA AND DEFINITIONS
Section 1.01. Basic Data and Definitions. The following sets forth basic data and, where appropriate, constitutes definitions of the terms hereinafter listed.
|
(a) |
“Property”The land and building (the “Building”) having an address at 440 US Highway 22, Bridgewater, New Jersey 08807. |
|
(b) |
“Permitted Use”For executive, general and administrative offices, training and any other lawfully permitted use consistent with a Class A office building, all subject to applicable Laws (as such term is hereinafter defined), and for no other use or purpose whatsoever without the prior written consent of Landlord. |
|
(c) |
“Premises”Approximately 67,747 rentable square feet consisting of the entire third floor of the Building, as measured in accordance with the “Standard Method for Measuring Floor Area in Office Buildings”, ANSI/BOMA Z65.1-2017 published by the Building Owners and Managers Association and as cross-hatched on Exhibit A attached hereto and made a part hereof. |
|
(d) |
“Commencement |
|
|
Date” |
Upon delivery of the Premises to Tenant in the Delivery Condition (as hereinafter defined) anticipated to be on or about July 1, 2019. |
|
(e) |
“Term”One Hundred Thirty-Two (132) months, commencing on the Commencement Date and ending on the Expiration Date (as hereinafter defined). |
|
(f) |
“Expiration Date”The last of the One Hundred Thirty-Second (132nd) month after the Commencement Date. |
|
|
|
(g) |
“Lease Year”As defined in Section 2.02. |
|
(h) |
“Base Rent” |
|
PERIOD |
BASE ANNUAL RENT |
BASE MONTHLY RENT |
|
* Month 1 through Month 12 |
$1,693,675.00 |
$141,139.58 |
|
Month 13 through Month 24 |
$1,727,548.50 |
$143,962.38 |
Page 2 of 60
|
$1,761,422.00 |
$146,785.17 |
|
|
Month 37 through Month 48 |
$1,795,295.50 |
$149,607.96 |
|
Month 49 through Month 60 |
$1,829,169.00 |
$152,430.75 |
|
Month 61 through Month 72 |
$1,863,042.50 |
$155,253.54 |
|
Month 73 through Month 84 |
$1,896,916.00 |
$158,076.33 |
|
Month 85 through Month 96 |
$1,930,789.50 |
$160,899.13 |
|
Month 97 through Month 108 |
$1,964,663.00 |
$163,721.92 |
|
Month 109 through Month 120 |
$1,998,536.50 |
$166,544.71 |
|
Month 121 through Month 132 |
$2,032,410.00 |
$169,367.50 |
* Notwithstanding anything contained herein to the contrary, provided Tenant is not then in default under the Lease beyond the expiration of any applicable cure period, Tenant shall be entitled to: (i) an abatement of rent against all Base Rent otherwise payable for the Premises for the first nine (9) months following the Commencement Date; (ii) an abatement of rent in the sum of $89,056.25 against the Base Rent otherwise payable for the Premises for Month 10 through Month 12; (iii) an abatement of rent in the sum of $90,837.38 against the Base Rent otherwise payable for the Premises for Month 13 through Month 15; (iv) an abatement of rent in the sum of $37,712.38 against the Base Rent otherwise payable for the Premises for Month 16 through Month 18; (v) an abatement of rent in the sum of $22,923.21 against the Base Rent otherwise payable for the Premises for Month 19 through Month 24; and (vi) an abatement of rent in the sum of $23,662.67 against the Base Rent otherwise payable for the Premises for Month 25 through Month 27. Tenant shall be responsible for all other sums and charges coming due and payable under this Lease during such period. For the purpose of clarity, provided Tenant is entitled to abatements set forth above, the following table shall calculate the amount of rent due after deducting the applicable abatements:
|
PERIOD |
BASE MONTHLY RENT |
ABATEMENT AMOUNT |
BASE RENT DUE AFTER ABATEMENT |
|
Month 1 through Month 9 |
$141,139.58 |
$141,139.58 |
$0.00 |
|
Month 10 through Month 12 |
$141,139.58 |
$89,056.25 |
$52,083.33 |
|
Month 13 through Month 15 |
$143,962.38 |
$90,837.38 |
$53,125.00 |
|
Month 16 through Month 18 |
$143,962.38 |
$37,712.38 |
$106,250.00 |
|
Month 19 through Month 24 |
$143,962.38 |
$22,923.21 |
$121,039.17 |
|
Month 25 through Month 27 |
$146,785.17 |
$23,662.67 |
$123,122.50 |
|
(i) |
“Security Deposit” $282,279.16 |
|
(j) |
“Tenant’s Proportionate |
|
Share”34%. |
|
(k) |
Renewal OptionsAs set forth in Section 2.04, Two (2) Five (5) year terms. |
|
(l) |
Right of First OfferAs set forth in Section 2.05. |
|
(m) |
Operating Expenses |
|
Base YearAs set forth in Section 9.01, Base Year 2019. |
|
(n) |
Electricity chargeAs set forth in Section 10.01. |
|
(o) |
Overtime HVACAs set forth in Section 10.03. |
Page 3 of 60
ARTICLE 2
DEMISE, TERM
Section 2.01. Demise. Landlord hereby demises and leases to Tenant, and Tenant leases, rents, and agrees to accept from Landlord, the Premises, upon the terms and conditions set forth in this Lease. The exterior walls and the roof (if applicable) of the Premises and/or to the bottom of the floor deck above the Premises, and the area beneath the Premises are not demised hereunder, and the use thereof, together with the right to locate, both vertically and horizontally, install, maintain, use, repair and replace columns, pipes, utility lines, ducts, conduits, flues, refrigerant lines, drains, sprinkler mains and valves, access panels, wires and structural elements leading through the Premises serving other parts of the Building, is reserved unto Landlord, so long as the same are located in and through the walls and ceilings of the Premises and do no reduce the rentable square footage of the Premises more than a de minimis amount. Landlord reserves an easement above Tenant’s finished ceiling (if any) to the roof and/or to the bottom of the floor deck above the Premises, as applicable, for general access purposes and in connection with the exercise of Landlord’s other rights under this Lease.
Section 2.02. Term and Lease Year. The Term shall commence on the Commencement Date and, unless sooner terminated or extended as provided in this Lease, shall terminate on the Expiration Date. The term “Lease Year” as used in this Lease shall mean the period of twelve (12) full calendar months commencing on the Commencement Date plus any partial month following the Commencement Date if the Commencement Date is not the first day of a month and each twelve (12) month period thereafter. Notwithstanding anything to the contrary contained herein, subject to Tenant Delay and force majeure, in the event the Premises has not been delivered in the Delivery Condition within one hundred fifty (150) days following the receipt of full permits for Phase One of the Landlord’s Work including finishing schedules (the “Outside Delivery Date”), Landlord shall provide Tenant with one (1) day of additional abatement of Base Rent for the Premises for each day the Premises is not delivered in the Delivery Condition beyond the Outside Delivery Date.
Section 2.03. Condition of Premises. Except for Landlord’s Work and except as otherwise expressly set forth herein, the Premises are leased to Tenant in their “as-is” condition with all faults and without representation or warranty by Landlord whatsoever, express or implied, as to the condition or suitably thereof or otherwise.
Section 2.04. Renewal Option. Provided that this Lease shall be in full force and effect and Tenant shall neither be in default hereunder after the expiration of the applicable notice or cure period at the time of its giving of notice of exercise nor on the commencement of the Renewal Term, Tenant shall have the option to renew this Lease (the “Renewal Option”) for two (2) additional periods of five (5) years each, commencing the day following the expiration of the initial Term (the “Renewal Term”) upon written notice to Landlord (“Tenant’s Exercise Notice”) on or before the date that is nine (9) months before the expiration of the Term. Time is of the essence with respect to Tenant’s giving of such notice. Upon the commencement of each Renewal Term, Base Rent for such Renewal Term shall be equal to Ninety-Five percent (95%) of the Fair Market Rent for a similar property in the Bridgewater, New Jersey market. Upon Tenant’s exercise of the Renewal Option, all references in this Lease to the Term hereof shall be deemed to mean the Term as so extended, except where expressly otherwise provided. For the purpose of this Section 2.04, “Fair Market Rent” shall mean the prevailing market rental rate for the Premises as of the first day of the applicable Renewal Term. If Landlord and Tenant cannot agree in writing upon the Fair Market Rent for the Premises within sixty (60) days of the delivery of Tenant’s Exercise Notice, then the Fair Market Rent will be determined pursuant to “baseball arbitration”, as follows:
(i) Landlord and Tenant shall each simultaneously present to the other party their final determinations of the prevailing market rate (the “Final Offers”) no later than sixty (60) days
Page 4 of 60
after the delivery of Tenant’s Exercise Notice. If the such rate as determined by the lower of the two (2) proposed Final Offers is not more than five percent (5%) below the higher, then such rate shall be determined by averaging the two (2) Final Offers.
(ii) If the difference between the lower of the two (2) proposed Final Offers is more than five percent (5%) below the higher, then within fifteen (15) days after each party has presented its Final Offer, the parties shall endeavor to select a mutually acceptable arbitrator (the “Arbitrator”), who shall be a qualified and impartial person licensed in the State of New Jersey as a commercial real estate leasing broker with at least ten (10) years of experience in the leasing of retail space comparable to the Premises in Somerset County, New Jersey. If Landlord and Tenant are unable to agree on the Arbitrator, either party, by giving ten (10) days' notice to the other party, can apply either to the New Jersey State Association of Realtors or to a Judge of the Superior Court of the County of Somerset for the selection of an arbitrator meeting the qualifications stated in this paragraph. The Arbitrator, however selected, shall be a person who has not previously acted in any capacity for either party. Each of the parties shall bear one-half of the cost of the Arbitrator and any fees of the court. Attorneys’ fees and expenses of counsel and of witnesses for the respective parties shall be paid by the respective party engaging such counsel or calling such witnesses.
(iii) The Arbitrator shall, after due consideration of the factors to be taken into account in determining the fair market rental rate for the Premises (and considered as though the Premises were vacant and unleased), and hearing whatever evidence the arbitrator deems appropriate from Landlord, Tenant and others, and obtaining any other information the arbitrator deems necessary, in good faith, make its own determination of the prevailing market rate for the Premises as of the first day of the applicable Renewal Term (the “Arbitrator's Initial Determination”) and thereafter select either Landlord's Final Offer or the Tenant's Final Offer, but no other, whichever is closest to the Arbitrator's Initial Determination (the “Final Determination”), such determination to be made within thirty (30) days after the appointment of the Arbitrator. The Arbitrator's Initial Determination, Final Determination and the market information upon which such determinations are based shall be in writing and counterparts thereof shall be delivered to Landlord and Tenant within said thirty (30) day period. The Arbitrator shall have no right or ability to determine the prevailing market rate in any other manner. The Final Determination shall be binding upon the parties hereto and shall be used to determine the Base Rent for the commencement of each Renewal Term.
Section 2.05. Right of First Offer. Subject to the rights of existing tenants as more particularly set forth on Schedule 2.05 attached hereto, Landlord hereby grants Tenant a right of first offer (“ROFO”) to lease space on the second (2nd) floor of the Building (the “ROFO Space”) if either (i) the ROFO Space first becomes available after the commencement of this Lease, or (ii) if ROFO Space that is vacant at commencement (or any portion thereof) becomes subject to an existing proposal from another tenant or perspective tenant which bona fide offer Landlord wishes to accept (“Existing Proposal”), as set forth below:
(a) If the ROFO Space first becomes available after the commencement of this Lease and there is not an Existing Proposal, Landlord shall give Tenant written notice to Tenant at such time as the ROFO Space becomes available for lease (such notice, “Landlord’s ROFO Notice”). Tenant shall have a period of two (2) business days from the date of Tenant’s receipt of the Landlord's ROFO Notice to give notice to Landlord that it desires to negotiate to lease all of the ROFO Space that is becoming available for lease. Except for the economic terms, Landlord and Tenant shall thereafter enter into a lease amendment for the ROFO Space upon the same terms as those found in this Lease; provided, however, the term for the ROFO Space shall be coterminous with the Term of this Lease, so long as there is at least five (5) years remaining in the Term, otherwise the Term of the Lease shall be extended such that the
Page 5 of 60
ROFO Space and the Premises are leased for a term no less than five (5) years. Once Tenant has provided Landlord with notice that it desires to negotiate to lease all of the ROFO Space that is becoming available, the parties shall thereafter negotiate in good faith those outstanding economic terms for the ROFO Space. In the event the parties are unable to negotiate such economic terms within a fifteen (15) day period, Tenant shall be entitled to rescind its notice to Landlord of its intention to negotiate to lease the ROFO Space and Landlord shall be entitled to lease all or such portion of the ROFO Space to any third party on any terms as Landlord shall see fit. If Tenant has not given notice to Landlord of its desire to negotiate within such two (2) business day period, or if Landlord and Tenant have not entered into a lease or lease amendment for all of the ROFO space becoming available within thirty (30) days of the determination by the parties of the economic terms for the ROFO Space, then Landlord shall thereafter be free to lease all or such portion of the ROFO Space to any third party on any terms as Landlord shall see fit; provided, however, in the event the ROFO Space becomes available again during the Term (as may be extended), Tenant shall be entitled to an additional Landlord’s ROFO Notice at such time as the ROFO Space becomes available thereafter in accordance with the terms and conditions of this Section 2.05.
(b)If the ROFO Space first becomes available after the commencement of this Lease and there is an Existing Proposal, or if ROFO Space that is vacant at commencement or any portion thereof becomes subject to an Existing Proposal, Landlord shall include the Existing Proposal along with Landlord’s ROFO Notice, and Tenant’s ROFO shall be limited to match the terms of the Existing Proposal. If Tenant has not given notice to Landlord of its desire to negotiate within such two (2) business day period, or, having given such notice, Landlord and Tenant have not entered into a lease or lease amendment for all or such portion of the ROFO space within thirty (30) days of the giving of Tenant’s notice of its desire to negotiate, then Landlord shall thereafter be free to lease the all or such portion of the ROFO Space to any third party on any terms as Landlord shall see fit; provided, however, in the event both (i) Landlord has not leased the ROFO Space subject to the Existing Proposal within sixty (60) days following Tenant’s receipt of Landlord’s ROFO Notice, and (ii) Landlord is not in active negotiations for the ROFO Space subject to the Existing Proposal, then Tenant shall be entitled to an additional Landlord’s ROFO Notice for such ROFO Space.
(c)In the event Landlord desires to market any available ROFO Space that is vacant as of the commencement of this Lease, but does not yet have an Existing Proposal, Landlord shall be obligated to provide Tenant with the terms upon which Landlord desires to market the ROFO Space (such notice, “Landlord’s Initial Offering”), and Tenant shall have two (2) business days to provide Landlord with notice of Tenant’s intention to negotiate to lease such ROFO Space. If Tenant has not given notice to Landlord of its intention to negotiate to lease such ROFO Space within such two (2) business day period, Landlord shall thereafter be free to lease all or such portion of the ROFO Space to any third party on any terms as Landlord shall see fit; provided, however, in the event an Existing Proposal is received, Tenant shall continue to have those rights as set forth in Section 2.05(b) above. Notwithstanding anything to the contrary contained herein, the parties hereby agree that Landlord may deliver the Landlord’s ROFO Notice or Landlord’s Initial Offering, as applicable, by email to the following email addresses: donna.pasek@amarincorp.com; michael.kalb@amarincorp.com (or such other email addresses as Tenant shall designate by notice given in accordance with Section 32.09) with the words “LANDLORD’S ROFO NOTICE” or “LANDLORD’S INITIAL OFFERING”, as applicable, in bold all-caps in the subject line of the email. Tenant shall be entitled to provide Landlord with Tenant’s response in reply to such email, all in accordance with this Section 2.05, and no additional written Notice shall be required by either Landlord or Tenant in order to effectuate the rights of either party under this Section 2.05.
ARTICLE 3
RENTAL
Section 3.01. Rental. The rents reserved under this Lease (collectively, the “Rental”), shall consist of Base Rent and all other sums as shall become due and payable by Tenant to Landlord under this
Page 6 of 60
Lease (collectively, “Additional Rent”). Tenant shall pay the Rental to Landlord at Landlord’s address first set forth hereinabove, or at such other place as Landlord may designate in writing from time to time, without any deduction, reduction, recoupment or set-off whatsoever and, except as otherwise expressly provided hereunder, without any statement whatsoever. If mailed, payment of all Rental shall be mailed in sufficient time and with adequate postage to be received by Landlord not later than the date due.
Section 3.02. Late Charge. If Tenant shall fail to pay when due any Rental within ten (10) days of the date due and payable, Tenant shall pay to Landlord, as Additional Rent, a late charge equal to five (5%) percent of the unpaid Rental, as an agreed and liquidated amount as compensation for Landlord’s additional administrative expenses relating to such late payment; provided, however, Tenant shall be entitled to a grace period of up to Five (5) days following written notice from Landlord to Tenant of such late payment for the first late payment of Rental in a calendar year. The provisions of this Section 3.02 is in addition to any other remedies available to Landlord with respect to non-payment of Rental.
ARTICLE 4
BASE RENT
Section 4.01. Base Rent. Commencing on the Commencement Date and continuing thereafter on the first day of each month throughout the Term, as and for its base rent (“Base Rent”) and subject to any abatements expressly set forth herein, Tenant shall pay to Landlord, in advance, without any notice or demand therefor, Base Rent set forth in the schedule set forth in Section 1.01(h). Base Rent shall be prorated on the basis of a thirty (30) day month for any partial month during the Term. Payment of the first month’s Base Rent is due upon the execution of this Lease.
ARTICLE 5
INTENTIONALLY OMITTED
ARTICLE 6
LANDLORD’S WORK
Section 6.01. Landlord’s Plans. Landlord’s Work shall be completed in two phases: (i) the first phase shall include approximately 50,000 rentable square feet (“Phase One”); and (ii) the second phase shall include the remaining approximately 17,747 rentable square feet (“Phase Two”). No later than thirty (30) days after the date of this Lease, Landlord shall cause Kimmerle Newman Architects (the “Architect”) to prepare and submit to Landlord construction drawings and specifications (“Construction Plans”) for the buildout of the Premises (“Landlord’s Work”) in accordance with the Phase One floor plans attached hereto as Exhibit B (the “Phase One Floor Plans”) and based on the Building Standards attached hereto as Exhibit B-1. During the Term of the Lease, Tenant may request that Landlord provide Construction Plans for the Landlord’s Work in accordance with the Phase Two plans attached hereto as Exhibit C (the “Phase Two Floor Plans”; the Phase One Floor Plans together with the Phase Two Floor Plans, the “Floor Plans”). With regard to both Phase One and Phase Two, the Construction Plans shall be subject to Tenant’s approval, not to be unreasonably withheld, conditioned or delayed so long as the same is consistent with the Phase One Floor Plans and Phase Two Floor Plans, as applicable. Tenant shall provide its written approval, or written denial together with specifications of all reasonable changes which would result in approval, within ten (10) business days of its receipt of the Construction Plans, failing which such Construction Plans shall be deemed approved by Tenant. In the event that Tenant timely submits its reasonably requested changes to the Construction Plans in accordance with the Floor Plans and Landlord approves of such changes, Landlord shall cause the Architect to make such changes and shall submit such revised Construction Plans to Tenant for its approval, not to be unreasonably withheld, conditioned or delayed. Tenant shall provide its written approval, or written denial together with specifications of all reasonable changes which would result in approval, within three (3) business days of its receipt of the revised Construction Plans, failing which such revised Construction Plans shall be
Page 7 of 60
deemed approved by Tenant. The foregoing process shall be repeated until the construction drawings have been approved (or deemed approved) by Tenant. The Construction Plans as approved or deemed approved by the parties are hereinafter referred to as the “Approved Construction Plans”. Notwithstanding anything to the contrary contained herein, the parties hereby agree that Landlord, as part of Phase One of Landlord’s Work, shall complete the restroom renovations on the third (3rd) floor. The design of such bathrooms shall be similar to that completed on the first (1st) floor of the Building, with the exception of adding hands free faucets, paper towel and soap dispensers. The design will not be included on the Phase One Floor Plans nor the Exhibit B-1 Building Standards, but shall be completed as part of Phase One of Landlord’s Work.
Section 6.02. Costs and Allowance. Landlord and Tenant acknowledge that the costs of designing and performing Landlord’s Work (including, without limitation, all soft and hard costs) shall be at Landlord’s sole cost and expense. Notwithstanding the foregoing, any costs relating to the performance of Landlord’s Work related to Change Orders (as hereinafter defined) shall be Tenant’s responsibility. For the avoidance of doubt, Landlord and Tenant acknowledge that it is the parties intention that the total cost for which Landlord is responsible for the Landlord’s Work for Phase Two shall be the same approximate cost on a per square foot basis as Landlord’s Work being completed for Phase One.
Section 6.03. Change Orders. If after the Construction Plans have been approved (or deemed approved), Tenant requests any revisions to the Approved Construction Plans and Landlord approves the revisions, Landlord shall cause Architect to revise the Construction Plans. After completion of the revisions, Landlord shall obtain from Landlord’s contractor and deliver to Tenant (a) an itemization of the increased cost (if any) in the Landlord’s Work resulting from such revisions to the Construction Plans, and (b) a reasonable estimate of the delay (if any) in the completion of the Landlord’s Work resulting from such revisions to the Construction Plans. Tenant, within three (3) business days of receiving such itemization, shall notify Landlord in writing whether it desires to proceed with such revisions. In the absence of such written authorization, Landlord shall have the option to continue work on the Premises disregarding the requested revisions. Any actual delay in completion of Landlord’s Work resulting from any revision or requested revision to the Construction Plans shall be deemed a Tenant Delay. If such revisions result in an increase in the cost of the Landlord’s Work in accordance with the foregoing, such increased costs, plus any applicable state sales or use tax thereon (collectively, the “Change Order Costs”), shall be payable by Tenant upon Landlord’s presentation of invoices therefor and as a condition to Landlord performing such Change Orders. In the event the Landlord performs any Change Orders before receiving payment from Tenant, and if Tenant fails to pay for the Change Order Costs within thirty (30) days of Landlord’s written demand (including reasonable backup for such Change Order Costs), then such Change Order Costs shall accrue interest at the Default Rate (as hereinafter defined) from the expiration of such thirty (30) day period until the date paid. Notwithstanding anything herein to the contrary, all revisions to the Construction Plans shall be subject to the prior approval of Landlord, which shall not be unreasonably withheld, delayed or conditioned. As used herein, “Change Orders” mean, collectively, any requested revisions to the Construction Plans and, if approved, corresponding changes to the Landlord’s Work, as applicable.
Section 6.04. Tenant’s Cooperation. Tenant agrees to fully cooperate with Landlord in order to enable Landlord’s Work to be performed in a timely manner. Notwithstanding anything herein to the contrary, any delay in the completion of Landlord’s Work or inconvenience suffered by Landlord during the performance of Landlord’s Work due solely as a result of a Tenant Delay shall not delay the Commencement Date nor shall it subject Landlord to any liability for any loss or damage resulting therefrom or entitle Tenant to any credit, abatement or adjustment of Rent or other sums payable under the Lease.
Section 6.05. Tenant Delay. The term “Tenant Delay” shall mean any actual delay in the completion of the Landlord’s Work by reason of any act or neglect, failure or omission (where there is a
Page 8 of 60
duty to act) of Tenant, its members, agents, servants, employees, contractors or subcontractors, or in the performance of Tenant's obligations under this Lease, including, but not limited to:
|
|
(i) |
Any delay due to Tenant’s failure to meet any Tenant performance deadlines set forth in this Lease; |
|
|
(ii) |
Any delay due to Change Orders requested by Tenant, regardless of whether or not such Change Order was approved by Landlord; |
|
|
(iii) |
Any interference by Tenant or any of its members, agents, servants, employees, contractors or subcontractors with the completion of Landlord’s Work; |
|
|
(iv) |
Tenant’s failure to pay when due the Change Order Costs; |
|
|
(v) |
Any delay of Tenant in making choices that need to be made by Tenant, if and to the extent applicable, which extend beyond either the time period specified in this Lease, or, in the event no time period is specified for such a decision, beyond three (3) business days following Landlord’s proposal thereof to Tenant, if any (and in the event of such failure, then, in addition to such failure constituting a Tenant Delay, Landlord shall have the right to perform Landlord’s Work with the inclusion of the particular specifications and choices as Landlord reasonably selects, and Tenant shall be deemed to have selected such specifications and choices); and |
|
|
(vi) |
Any default of Tenant under this Lease beyond applicable notice and cure periods. |
Notwithstanding anything to the contrary contained in the Lease, upon the occurrence of a Tenant Delay, the Commencement Date shall be accelerated and shall be deemed to occur on the date when the Premises would have been ready for delivery as required herein but for such Tenant Delay. Notwithstanding anything to the contrary contained herein, except where a Tenant Delay arises from Tenant’s failure to timely act on or before a date or time period expressly set forth in the Lease (in which event no Tenant Delay Notice shall be required): (y) in no event shall any act or omission be deemed to be a Tenant Delay until and unless Landlord has given Tenant written notice, which may be provided by email to the following email addresses donna.pasek@amarincorp.com; michael.kalb@amarincorp.com; and Lori.Stanton@am.jll.com (the “Tenant Delay Notice”) advising Tenant (i) that a Tenant Delay is occurring, and (ii) of the basis on which Landlord has determined that a Tenant Delay is occurring, and (z) no period of time prior to the time that Tenant receives a Tenant Delay Notice shall be included in the period of time charged to Tenant pursuant to such Tenant Delay Notice. Such Tenant Delay Notice, if given by email shall include the following in the subject line in ALL CAPS: “REGARDING TENANT DELAY”. For the avoidance of doubt, Landlord shall be entitled to claim a Tenant Delay from the day the Tenant Delay Notice is delivered to Tenant in accordance with this Section 6.05.
Section 6.06. Delivery Condition. Landlord shall cause Landlord’s Work to be constructed in compliance with applicable Laws and building code requirements, and in a good and workmanlike manner. Upon the Commencement Date, Landlord shall deliver the Premises in broom clean condition, with all mechanical systems, bathrooms, entrances, sidewalks, roof, and structure in good working order. As used in this Lease, the term “Delivery Condition” means the substantial completion of Landlord’s Work. Landlord’s Work shall be deemed substantially complete when it is completed in accordance with this Lease, except for minor details of construction, decoration and mechanical adjustments to be performed by Landlord, the noncompletion of which does not materially interfere with Tenant’s performance of its Tenant Work (as hereinafter defined). Upon notification by Landlord after the substantial completion of Landlord’s Work, Landlord and Tenant shall schedule a pre-occupancy inspection of the Premises at which time a punchlist of outstanding items, if any, shall be
Page 9 of 60
completed. Within a reasonable time thereafter, Landlord shall complete the punchlist items to Tenant’s reasonable satisfaction. Tenant’s acceptance of the Premises shall be deemed to be an acceptance of the satisfactory completion of Landlord’s Work, subject only to Landlord’s completion of the punchlist items identified by Tenant in writing during such inspection. Landlord does not warrant Landlord’s Work whatsoever. Notwithstanding anything to the contrary contained herein, provided, and upon condition that Tenant shall give written notice to Landlord of such defect, Landlord shall warranty the Landlord’s Work for (i) ninety (90) days from the date on which Tenant takes possession of the Premises as to patent defects; and (ii) one (1) year from the date on which Tenant takes possession of the Premises as to latent defects. To the extent any components of Landlord’s Work are covered by a manufacturer’s or contractor’s warranty running to Landlord’s benefit, Landlord shall use reasonable efforts to enforce the terms of such warranty such that Tenant receives the benefit thereof.
Section 6.07. Tenant’s Finish Work. All finish work and decoration and other work desired by Tenant and not included within the Landlord’s Work as set forth in the approved Construction Plans including, without limitation, all computer systems, cabling, telephone systems, telecommunications systems, fixtures, furnishings, furniture and equipment (collectively, the “Tenant Work”) will be designed, furnished and installed by Tenant at Tenant’s sole expense and shall be deemed Tenant’s Changes (as such term is defined in Article 12) and shall be subject to the terms of Article 12. Tenant shall ensure that Tenant’s Work shall not exceed the floor load of the Premises.
Section 6.08.Early Access. Concurrent with Landlord performing Landlord’s finish work, but no later than four (4) weeks prior to the estimated Commencement Date, provided that Tenant shall not interfere with Landlord’s Work, Landlord shall permit Tenant to enter the Premises and Common Areas, if necessary, in order to commence installing its equipment, racking system, cabling, wiring, fixtures, and furniture, subject to Tenant obtaining, at Tenant's sole cost and expense, all permits in connection with the installation thereof. With respect to such early access, all provisions of this Lease shall then be in full force and effect, specifically including, but not limited to, Articles 7 and 14 hereof (excluding however, Tenant's obligation to pay Base Rent or Additional Rent, including electricity).
ARTICLE 7
USE AND OPERATION OF THE PREMISES
Section 7.01. Use. Tenant shall use the Premises for the Permitted Use and for no other use or purpose whatsoever. Tenant shall have access to the Premises, Building, and parking lot, 24 hours per day, 7 days per week, 52 weeks per year.
Section 7.02. Compliance With Laws. Subject to Landlord’s obligation to deliver the Premises in compliance with all applicable Laws, Tenant, at Tenant’s sole cost and expense, shall promptly comply with all present and future laws, statutes, ordinances, codes, rules and regulations of any governmental or quasi-governmental authority having jurisdictions over the Premises (collectively, “Laws”) affecting or applicable to (i) Tenant’s manner of use of the Premises (as opposed to general office, administrative or training purposes) or Tenant’s Changes (as defined in Article 12), or (ii) Tenant’s business conducted on the Premises, whether or not any such Laws are foreseen or unforeseen, ordinary or extraordinary, or shall interfere with the use and enjoyment of the Premises; provided, however, that such compliance shall be required by virtue of Tenant’s manner of use of the Premises (as opposed to general office, administrative or training purposes) or Tenant’s Changes. Upon Landlord’s written request, Tenant shall deliver to Landlord true and complete copies of any and all permits, licenses and/or certificates required for the lawful conduct of Tenant’s business in the Premises. Notwithstanding the foregoing or anything contained herein to the contrary, Landlord shall be responsible for compliance with the ADA and all Laws regarding accessibility to the Premises. Landlord hereby represents that the Building meets all current ADA standards and requirements as of the date of this Lease.
Page 10 of 60
Section 7.03. Compliance With Insurance Requirements. Other than Landlord’s Work as set forth in Section 6.01, Tenant, at Tenant’s sole cost and expense, shall comply with any and all provisions, recommendations and requirements of (i) any national or local Board of Fire Underwriters (or other similar body) having jurisdiction over any part of the Premises and (ii) any insurance policy(ies) covering or applicable to the Premises or Building and any issuer(s) of such insurance policy(ies) (collectively, “Insurance Requirements”); provided, however, that such Insurance Requirements relate to Tenant’s manner of use of the Premises not otherwise allowed under the Permitted Use or Tenant’s Changes.
Section 7.04. Manner of Use. In no event shall the Premises or any portion thereof be used: (i) in violation of any Laws, Insurance Requirements or the certificate of occupancy or other licenses or certificates covering the Premises; (ii) in a manner which creates or permits a nuisance or trespass; (iii) in a manner which produces, reproduces, or transmits sounds audible outside the Premises; (iv) in a manner which obstructs or encumbers the Common Areas (as hereinafter defined); (v) in a hazardous manner; (vi) in a manner which exceeds the floor load which such floor was designed, or is permitted by Laws, to carry; (vii) to display or operate vending machines or coin or token operated amusement devices except for employees’ use only; (viii) in any manner which causes or permits any noise, odors, fumes, dust or vapors to emanate or to be dispelled from the Premises; provided, however, nothing in this Section 7.04 is intended to prohibit Tenant from using the Premises for the Permitted Use.
Section 7.05. Hazardous Substances. (a)Tenant shall not engage in operations at the Premises which involve the generation, manufacture, refining, transportation, treatment, storage, handling or disposal of any “hazardous substance” or “hazardous waste” as such terms are defined under the Industrial Site Recovery Act, N.J.S.A. 13"1K-6 et seq. (West Supp. 1989) (“ISRA”), or of “hazardous substances” as defined in section 101 (14) of the Comprehensive Environmental Response, Compensation and Liability Act (“CERCLA”), 42 U.S.C. 9601 (14), as amended by the Superfund Amendments and Reauthorization Act of 1986 (“SARA”).
(b)Upon written request of Landlord, Tenant shall cooperate with Landlord in obtaining evidence of compliance with ISRA or with any other law, regulation, or order of any governmental authority, which cooperation shall include, without limitation, providing affidavits, reports or responses to questions.
(c)Tenant represents that its North American Industrial Classification System (“NAICS”) number, as designated in the Standard Classification Manual prepared by the Office of Management and Budget, Executive Office of the President of the United States, is 32541. Tenant recognizes that for purposes of ISRA, Tenant will acquire the NAICS number of any entity for which it provides all or substantially all of its services or products.
(d)Tenant represents and warrants that the specific activities intended to be conducted on the Premises are in accordance with provisions in this Section 7.05 and will not constitute an Industrial Establishment subject to the requirements of ISRA.
(e)Tenant further covenants that from and after the Commencement Date it will not cause or permit to exist, as a result of an intentional or unintentional action or omission on its part, the releasing, spilling, leaking, pumping, pouring, emitting, emptying or dumping on or about the Premises of any “hazardous substance” as such term is defined in N.J.S.A. 58:10-23.11b (k) and N.J.A.C. 7:1-3.3).
(f)If Tenant, in violation of the foregoing covenants, ever causes through an act or omission of Tenant or anyone claiming by, through or under Tenant, the Premises to become an Industrial Establishment, then Tenant shall:
Page 11 of 60
(i)indemnify, defend and hold harmless Landlord, its successors and assigns, and any partner and any other agent or employee of Landlord, or its successors or assigns (collectively, the “Indemnified Parties”), from and against any and all claims, demands, actions, causes of actions, suits, judgments, damages, losses, liabilities, fines, penalties, costs and expenses (including, without limitation, reasonable attorneys’ fees and disbursements) (collectively, “Liabilities”) resulting from, arising out of, or in any way connected with injury to or the death of any person (including any Indemnified Party) or physical damage to property of any kind wherever located and by whomever owned (including any Indemnified Party) arising out of or in any way connected with the presence on, in or under the Premises of any asbestos, polychlorinated biphenyls (PCB's) or any “hazardous substance” or “hazardous waste” as such terms are defined by ISRA, CERCLA or the Spill Act (as hereinafter defined) or of any other hazardous, toxic or polluting substance or material; and
(ii)at its expense, comply with all applicable requirements of ISRA pertaining to the transfer or closure of an Industrial Establishment. Without limitation of the foregoing, Tenant understands that is obligations might include (1) the proper filing of an initial notice to the New Jersey Department of Environmental Protection (the “DEP”), (2) the performance of any soil, groundwater and surface water sampling and test required by the DEP, and (3) either the filing of a “negative declaration” with the DEP or the performance of a proper and approved cleanup plan to the satisfaction of the DEP. Further, in the event Landlord enters into an agreement to sell the Premises at any time during the Term, Tenant will, upon request, and at Landlord's expense, furnish to Landlord such statements, affidavits and information as Landlord may require in order to file an initial notice and thereafter obtain a negative declaration. In the event that the DEP requires the implementation of a cleanup plan prior to issuance of a negative declaration, which plan is required due to Tenant's operations at the Premises, then Tenant shall bear the cost implementation of the cleanup plan.
(g)Tenant shall not operate a “major facility” at the Premises within the meaning of the New Jersey Spill Compensation and Control Act (the “Spill Act”), N.J.S.A. 58:10-23.11 to 58:10-23.11z (West 1982 and Supp. 1989) as such law may be amended from time to time. Tenant has no knowledge of any lien imposed upon its revenues, real or personal property pursuant to Section 7 (f) of the Spill ACT, N.J.S.A. 58:10-23.11f (f) (West Supp. 1989), as such section may be amended from time to time, and Tenant has no actual or constructive knowledge of any circumstances which might lead to the imposition of such a lien.
(h)In the event of Tenant's failure to comply in full with this Section 7.05, Landlord may, at is option, perform any and all of Tenant's obligations as aforesaid and all reasonable costs and expenses incurred by Landlord in the exercise of this right shall be deemed to be Additional Rent payable upon demand.
(i)Except to the extent caused by the actions of Tenant or its agents, employees or contractors, Landlord will indemnify, defend and hold Tenant harmless from all losses, damages and expense incurred by Tenant as a result of any Hazardous Substances that have been released or emitted within the Property prior to Tenant’s access, occupancy, or possession of the Premises. Landlord represents that there are no environmental violations in or around the Building that pose a present danger to health, life or safety including, but not limited to, the presence of asbestos, radon, PCBs and mold.
(j)This Section 7.05 shall survive the expiration or some determination of this Lease.
ARTICLE 8
COMMON AREA
Section 8.01. Non-exclusive License. Tenant, its employees, agents, contractors and invitees, shall have a non-exclusive license, in common with other tenants of the Building and their employees,
Page 12 of 60
agents and invitees, to use the parking areas (subject to the conditions set forth herein) and any areas which may from time to time be made available for the joint use of tenants of the Property (“Common Area”), for their intended purposes. Landlord does not warrant the continuing layout or configuration of the Property (including the Common Area), and Landlord shall have the unrestricted right to construct from time to time additional improvements on the Building or increase, reduce, eliminate, relocate or change the size, dimensions, design, configuration or location of any or all of the Common Area (including, without limitation, the parking area) or other improvements in the Building in any manner whatsoever. Landlord represents that the following improvements to the Building in which the Premises is located are anticipated to be completed in the first quarter of the 2020 calendar year:
|
|
• |
Common conference space, |
|
|
• |
Fitness facilities. |
Notwithstanding anything to the contrary contained herein, the parties acknowledge that the operation of the café at the Building was a significant inducement to Tenant entering into this Lease and it is the intention of the Landlord to operate the café and/or some other appropriate active food service during the entire Term of this Lease. In the event (i) the café in the Building is operating at a net loss for a period longer than six (6) months, and (ii) Landlord intends to stop the operation of the café for more than a temporary period or as the result of a casualty, Landlord shall provide Tenant with at least ninety (90) days advance notice of Landlord’s intention to terminate the operation of the café. In the event the café is overburdened in its service to the tenants of the Building, Landlord shall make commercially reasonable efforts to ease such overburdening (such as introducing a grab and go kiosk, food cart, or adding additional capacity to the café, etc.), in order to have the café service the tenants within the Building in an efficient and effective manner.
Section 8.02. Common Area Definition. Landlord shall maintain or cause to be maintained the Common Area. The term “Common Area” shall include, as constituted and designated by Landlord from time to time: (i) all areas and space provided by Landlord for the common or joint use and benefit of tenants in the Building and/or on the Property (including any expansion thereof to adjacent and contiguous land), their employees, agents and invitees, including, without limitation, parking areas, access roads, driveways, retaining walls, landscaped areas, truck serviceways or tunnels, pedestrian walks, outside courts and curb cuts, and (ii) all other non-leasable portions of the Building.
ARTICLE 9
OPERATING EXPENSES
Section 9.01. Payment of Operating Expenses. In addition to the Base Rent payable hereunder, as set forth in Section 1.01(h), Tenant shall pay to Landlord, as Additional Rent, Tenant’s Proportionate Share of the amount by which the Operating Expenses in any calendar year exceed the Operating Expenses for the 2019 calendar year (the “Base Amount”), subject to adjustments as set forth in this Article 9.
Section 9.02. Tenant’s Payment. Commencing on the Commencement Date, and continuing on the first day of each calendar month in advance during the Term, Tenant shall pay to Landlord, as Additional Rent, such amount as Landlord shall reasonably estimate to equal one-twelfth (1/12th) of Tenant’s Proportionate Share of an amount equal to (x) estimated Operating Expenses for the then current calendar year, less (y) the Base Amount (the “OpEx Escalation”). If Operating Expenses due for any calendar year during the Term shall be due for a period of less than twelve (12) months, the Base amount and the amount of the OpEx Escalation for such calendar year shall be appropriately prorated. Notwithstanding the foregoing or anything to the contrary contained herein, Tenant shall not be responsible to pay the OpEx Escalation for the first 12 months after the Commencement Date.
Page 13 of 60
Section 9.03. Year End Adjustment. After the expiration of each calendar year, Landlord shall determine the total actual Operating Expenses for such calendar year, together with Tenant’s Proportionate Share thereof and shall forward to Tenant a detailed statement of Operating Expenses and calculation of Tenant’s Proportionate Share thereof. If the amounts paid by Tenant for such preceding calendar and/or fiscal year are less than Tenant’s Proportionate Share thereof, the deficiency shall be paid by Tenant to Landlord within thirty (30) days of written demand therefor; if Tenant shall have previously paid in excess of Tenant’s Proportionate Share, the amount of such excess shall be credited to the next Rental payable under this Lease, unless if such payment has been made with respect to the last Lease Year of the Term, in which case Landlord shall refund the amount of such overpayment to Tenant, provided Tenant is not then in default beyond any applicable notice or cure period. Landlord’s failure to render or delay in rendering any statement with respect to Operating Expenses shall not prejudice Landlord’s right to thereafter render such a statement(s) nor shall the rendering of such statement(s) for any calendar year prejudice Landlord’s right to thereafter render a corrected statement(s) for such calendar year. Tenant’s obligation to pay the OpEx Escalation for the last (or any earlier) calendar year during the term of this Lease shall survive the expiration or earlier termination of this Lease. Notwithstanding anything to the contrary contained herein, Tenant shall have no liability for any Operating Expenses not billed to Tenant by Landlord within two (2) years of the occurrence of such expense. No later than sixty (60) days following Tenant’s receipt of the Statement, Tenant shall have the right, during regular business hours and after giving Landlord at least twenty (20) days' advance written notice given during such sixty (60) day period, to complete an inspection or audit, or cause its agents to complete an inspection or audit, of Landlord's books and records relating to Operating Expenses for the immediately preceding calendar year. Such inspection or audit (hereinafter referred to as “Tenant's Audit”) shall take place at a mutually convenient date and time, at any of Landlord's office locations in New Jersey. Tenant’s Audit may not be performed on a contingency fee basis. If Tenant's Audit shows that the amounts paid by Tenant to Landlord on account of Operating Expenses exceed the amounts to which Landlord is entitled hereunder, then Landlord shall either credit such excess toward Tenant's next monthly Rent payment or promptly refund such excess to Tenant, and if Tenant’s Audit shows that the amounts paid by Tenant to Landlord on account of Operating Expenses is less than the amounts to which Landlord is entitled hereunder, then Tenant shall pay such deficiency to Landlord together with its next payment of regular Rental. In the event Tenant’s Audit shows that the amounts paid by Tenant to Landlord on account of Operating Expenses exceeds the amounts to which Landlord is entitled by more than ten percent (10%), then Landlord shall be obligated to pay the reasonable out of pocket expenses for Tenant’s Audit, in an amount not to exceed $7,500.
Section 9.04. Operating Expenses. As used in this Article 9, “Operating Expenses” shall mean the following expenses incurred by Landlord in connection with the operation, repair and maintenance of the Building:
|
|
(a) |
Real estate taxes and other taxes or charges levied in lieu of such taxes, general and special public assessments, charges imposed by any governmental authority pursuant to anti-pollution or environmental legislation, provided same are not the result of any act of failure to act on the part of the Landlord, any predecessor of Landlord, or any other tenant of the Building, taxes on the rentals of the Building or the use, occupancy or renting of space therein, together with reasonable costs incurred in connection with proceedings brought to contest the real estate taxes; |
|
|
(b) |
Commercially reasonable premiums and fees for insurance, including, but not limited to, fire and extended coverage insurance, insurance against loss of rentals for space in the Building and public liability insurance, all in amounts and coverages (with additional policies against additional risks) as may reasonably be required by Landlord or the holder, now or hereafter, of any mortgage on the Building; |
Page 14 of 60
|
|
(d) |
Maintenance and repair costs, including, but not limited to, repairs and replacements described in Section 11.01, repairs and replacements of supplies and equipment, snow removal and trash removal (including, without limitation, all costs associated with the separation and recycling of trash and other disposable materials) and paving, parking lot, walkways, lawn and general grounds upkeep, maintenance and repair, and the costs of all labor, material and supplies incidental thereto, except to the extent such repair is necessitated by the negligence or willful misconduct of the Landlord, is agents, employees and contractors; |
|
|
(e) |
Wages, salaries, fees and other compensation and payments and payroll taxes and contributions to any social security, unemployment insurance, welfare, pension or similar fund and payments for other fringe benefits required by law, union agreement or otherwise made to or on behalf of all employees of Landlord performing services rendered in connection with the operation, cleaning and maintenance of the Building and/or Land, including, without limitation, payments made directly to or through independent contractors for performance of such services; |
|
|
(f) |
Reasonable management fees payable to the managing agent for the Building provided that at no time shall the management fees exceed six percent (6%) of the collected gross rent of the Building on a monthly basis. As used herein, the term “gross rent” shall mean the amount of annual base rent collected under the leases and any operating expenses, taxes and utilities paid by tenants, in all cases excluding the security deposits; |
|
|
(g) |
Assessments paid by Landlord in respect of the repair, maintenance and upkeep of common facilities located in the Building; |
|
|
(h) |
The cost of any capital improvements made (i) for the primary purpose of reducing operating expenses or (ii) which may be required by governmental authority under any governmental laws or regulations first in effect after the date of this Lease (collectively, the “Permitted Capital Improvements”), which Permitted Capital Improvements shall be amortized over the useful life of such Permitted Capital Improvements in accordance with generally acceptable accounting principles, together with interest on the unamortized balance at the rate equal to the announced “prime rate” then in effect at Citibank, N.A., New York, New York (or its successor) on short-term unsecured loans to its largest and most credit worthy commercial customers, or such higher rate as may have been paid by Landlord on funds borrowed for the purpose of constructing such capital improvements; |
|
|
(i) |
Any and all other expenditures of Landlord incurred in connection with the operation, repair or maintenance of the Premises, or the Building which are properly expensed in accordance with generally accepted accounting principles consistently applied in the operation, maintenance and repair of a first-class office building facility. |
Notwithstanding anything to the contrary herein, the Base Amount and all Operating Expenses that vary by occupancy shall be adjusted to reflect 95% Building occupancy. Notwithstanding anything to the contrary contained herein, in no event shall Landlord be entitled to more than one hundred percent (100%) of the total cost of Operating Expenses for any operating year.
Page 15 of 60
Notwithstanding anything to the contrary contained herein, in no event shall the definition of Operating Expenses include the following: (i) legal or other professional fees related to leasing, ownership, financing, tenant disputes or other services not related to the normal operation, maintenance, cleaning, repair or protection of the Property; (ii) the cost of any additions, changes, replacements, painting, decorating, renovations and other items that are made solely in order to prepare tenant space for a new tenant’s occupancy; (iii) depreciation and interest and principal payments on mortgages and other debt costs, if any, other than amortization of and the interest factor attributable to Permitted Capital Improvements; (iv) costs and expenses associated with the operation of the business of the person or entity which constitutes Landlord as the same are distinguished from the costs of operation of the Property, including accounting and legal matters with respect to same; (v) costs of selling or financing any of Landlord’s interest in the Property; (v) income, excess profits, franchise taxes or other such taxes imposed on or measured by the net income of Landlord from the operation of the Building; (vi) costs incurred by Landlord for the repair of damage to the Building to the extent that Landlord is reimbursed by insurance proceeds; (vii) the costs of special services and utilities separately chargeable to individual tenants of the Building; (viii) intentionally omitted; (ix) marketing costs, brokerage commissions and concessions and leasehold improvement costs incurred in connection with the leasing of any rentable space at the Property, including, without limitation, finders’ fees, attorneys’ fees and expenses, entertainment costs and travel expenses; (x) accountants’ fees incurred in connection with disputes with individual tenants and/or the existence, maintenance or non-Property related operations of the legal entity or entities of which Landlord is comprised; (xi) the cost of any special work or service performed for any tenant (including Tenant) billable to such tenant or any costs in connection with services or benefits that are provided to or for the particular benefit of other tenants but not offered to Tenant; (xii) any expenses for repairs or maintenance actually covered by warranties and service contracts; (xiii) any cost that Tenant pays for directly; (xiv) costs of developing and constructing the other improvements or additions at the Building or Property, whether capital expenditures or otherwise; (xv) capital repairs or replacements of the façade, structural columns, floor slabs, structural portions of the roof (including any roof slab), or foundations of the Property (regardless of whether or not caused by latent defects), unless on account of changes in applicable which first become effective after the date of this Lease, and unless on account for capital expenses for improvements or building elements added to the Building which in Landlord’s reasonable judgment will increase the efficiency of the Building (i.e., are reasonably anticipated by Landlord to reduce the Operating Expenses as they relate to the item which is the subject of the capital expenditure or reduce the rate of increase in the Operating Expense which relates to the item which is the subject of the capital expenditure from what it otherwise may have been reasonably anticipated to be in the absence of such capital expenditure); (xvi) intentionally omitted; (xvii) the cost of removing or remediating hazardous materials from the Property; (xviii) any cost covered by any warranty that Landlord actually obtains in connection with the Property; (xix) any amounts paid to a person, firm, corporation or other entity under common ownership and control with Landlord that is in excess of a commercially reasonable amount paid on a market rate basis; (xx) the cost of acquiring sculptures, paintings and other objects of art; (xxi) the cost of advertising or promotion for the Property or any part thereof or any operations at the Property; (xxii) depreciation of the Property or any part thereof; (xxiii) accounting and bookkeeping services to the extent not allocable to the Property; (xxiv) any compensation paid to personnel in retail concessions operated by Landlord and any subsidies or concessions to third parties operating retail concessions at the Property; (xxv) salaries and bonuses and benefits of officers, executives of Landlord and administrative employees above the grade of property manager or building supervisor; (xxvi) Landlord’s general overhead; (xxvii) lease payments for rental equipment that would constitute a major capital expenditure if the equipment were purchased, but only to the extent that the capital expenditure itself would be excluded from Operating Expenses if the equipment were purchased; (xxviii) replacement or contingency reserves; (xxix) all capital improvements or expenditures, except for Permitted Capital Expenditures; (xxx) charges for the capital replacement and capital repairs of building foundation, structure, exterior walls and roof; (xxxi) any costs incurred with respect to the retail portions of the Building; and (xxxii) items for which Landlord is actually reimbursed by tenants (other than through general operating expense provisions) or other third parties.
Page 16 of 60
ARTICLE 10
UTILITIES AND SERVICES
Section 10.01. Electricity. (a) Landlord will furnish to Tenant alternating electric current to be used by Tenant for the lighting fixtures and electrical receptacles installed in the Premises, but Landlord shall not be liable in any way to Tenant for any failure or defect in supply or character of electric current furnished to the Premises except for the negligence or willful misconduct of Landlord and its agents, employees or contractors. Tenant's use of electric current in the Premises shall not at any time exceed the capacity of any of the electrical conductors and equipment in or otherwise serving the Premises. Tenant shall not make or perform, or permit the making or performing of, any alterations to wiring installations or other electrical facilities in or serving the Premises or any material additions to the machines, equipment and other appliances in the Premises without the prior written consent of Landlord in each instance, which shall not be unreasonably withheld, conditioned or delayed. Should Landlord grant any such consent, all additional risers or other equipment required therefor shall be installed by Landlord and the actual cost to Landlord thereof shall be paid by Tenant, within thirty (30) days of Landlord’s written demand, as Additional Rent. Tenant shall not overload the electrical capacity of the Premises as it exists on the Commencement Date.
(b) Commencing on the Commencement Date, in addition to the Base Rent, for and in consideration for the electrical services provided by Landlord under Section 10.01(a), Tenant shall pay, as Additional Rent, the annual sum of One Hundred Eighteen Thousand Five Hundred Fifty-Seven and 25/100 Dollars ($118,557.25) (i.e. $1.75 per rentable square foot), which shall be payable in equal monthly installments of Nine Thousand Eight Hundred Seventy Nine and 77/100 Dollars ($9,879.77) in advance on the first day of each month, and shall be deemed additional rent payable under this Lease; provided, however, that the foregoing rate shall be subject to increase based on: i) rate increases by the applicable utility company above the rate of the applicable utility company at the time of this Lease; and ii) an audit conducted on the Tenant’s electricity consumption, in which case Tenant shall pay for Tenant’s over-consumption of electricity based on the rate of the applicable utility company. Notwithstanding the foregoing, (i) if in Landlord’s reasonable discretion, Tenant equipment consumes electricity in an amount that exceeds $1.75 per rentable square foot of the Premises, Landlord shall have the right, at Landlord’s sole cost and expense, to install submeters separately measuring Tenant’s consumption of electricity at the Premises and Tenant shall pay to Landlord, within thirty (30) days of Landlord’s written demand, the actual costs of Tenant consumption of electricity at the Premises; and (ii) Tenant, may request that Landlord, at Landlord’s sole cost and expense, install submeters separately measuring Tenant’s consumption of electricity at the Premises and Tenant shall pay to Landlord, within thirty (30) days of Landlord’s written demand, the actual costs of Tenant consumption of electricity at the Premises.
Section 10.02. Water, Sprinkler and Fire Alarm. Landlord shall provide hot and cold water to the bathrooms in the common areas of the Building and provide sprinkler and fire alarm service for the Building.
Section 10.03. Heating, Ventilating and Air Conditioning Services. The heating, ventilating and air conditioning systems existing in the Premises as of the date hereof (the “HVAC System”) include a year-round air conditioning system during regular Business Hours sufficient to provide the following: (i) an interior ambient temperature of no more than 75°F when outside conditions are not in excess of 92°F DB, and (ii) heating with the heat output controlled by inside air temperature to maintain interior ambient air temperature of no less than 72°F DB when outside air temperature is 10° F or more. Maintained temperatures shall be as above unless otherwise required by law or governmental guidelines and subject to Tenant’s obligations set forth herein, and Tenant shall not set the heat to provide for temperatures above or below the temperatures set forth in the foregoing sentence. The proper performance of the HVAC System serving the
Page 17 of 60
Premises is based upon (A) a maximum density of one person per 150 rentable square feet, (B) a maximum electric heat gain of 4 watts per net usable square foot, (C) Tenant at all times (even during non-business hours) keeping the vents open and the base boards at a setting sufficient to maintain temperatures of not more than 78 degrees F during the cooling seasons and not less than 60 degrees F during the heating seasons, and (D) excess heat generated by Tenant’s equipment. Landlord shall not be responsible for the proper performance of such HVAC system if the Premises (or any room or area thereof) shall be subjected to a greater population density or a greater heat gain than above specified (whether due to the installation of additional equipment by Tenant or otherwise), if the partitioning in the Premises shall be rearranged in such manner as to interfere with the normal operations of the HVAC system in the Premises, if the windows and the public corridor entrance doors of the Premises shall not be kept closed, if the blinds shall not be lowered in windows of the Premises when exposed to the sun, or if Tenant shall otherwise fail to operate the HVAC system as required above. Landlord shall have free and unrestricted access to all HVAC equipment located in or accessible through the Premises. For the purpose of this Section 10.03, “Business Hours” shall mean Monday through Friday 8AM to 6PM and Saturdays 8AM to 1PM, excluding legal holidays. If Tenant requires after hours HVAC, such HVAC shall be available to Tenant upon not less than twenty-four (24) hours’ prior written request to Landlord, such written request may be emailed to info@broadmgmt.com or entered into the management company’s work order software, for an additional cost at Landlord’s then current rate which as of the date of this Lease is Eighty-five and 00/100 Dollars ($85.00) per hour, with a minimum requirement of four (4) hours per each request by Tenant. Notwithstanding the foregoing, Tenant shall receive an annual credit of one hundred (100) hours of after hours HVAC, with a minimum requirement of four (4) hours per each request by Tenant.
Section 10.04. Cleaning. Landlord shall furnish janitorial services and materials as required, consistent with a Class A office building in the Bridgewater/Somerset County market, for the reasonable occupancy and use of the Premises; provided, however, that Landlord shall not be required to provide janitorial services and trash removal to the Premises and the Common Areas of the Building more than five (5) time per week. Landlord shall furnish janitorial services and materials to the restrooms serving the Premises on such days as the Building is fully operational.
Section 10.05. No Liability. Landlord shall have no liability to Tenant or any other party for any inadequacy, cessation, or interruption of any service provided under this Article 10. Notwithstanding anything contained in this Lease to the contrary, if (i) an interruption or curtailment, suspension or stoppage of an Essential Service (as said term is hereinafter defined) shall occur as a result the negligence or willful misconduct of Landlord, its agents, contractors, or employees, except any of the same due to any act or neglect of Tenant or Tenant’s agents employees, contractors or invitees or any person claiming by, through or under Tenant (any such repair, negligence, or willful misconduct, or interruption of an Essential Service being hereinafter referred to as a “Service Interruption”), and (ii) such Service Interruption continues for more than three (3) consecutive business days after Landlord shall have received notice thereof from Tenant, and (iii) as a result of such Service Interruption, the conduct of Tenant’s normal operations in the Premises are materially and adversely affected, then there shall be an abatement of one day’s Rental for each day during which such Service Interruption continues after such three (3) business day period; provided, however, if the entire Premises have not been rendered unusable by the Service Interruption, the amount of abatement shall be equitably prorated. For purposes hereof, the term “Essential Services” shall mean the following services: access to the Premises, water and sewer/septic service and electricity, air conditioning and heating. Any abatement of rent under this paragraph shall apply only with respect to rent allocable to the period after each of the conditions set forth in subsections (i) through (iii) hereof shall have been satisfied and only during such times as each of such conditions shall exist.
ARTICLE 11
MAINTENANCE AND REPAIRS, SURRENDER OF POSSESSION
Page 18 of 60
Section 11.01. Landlord’s Obligations. Except as provided in Article 15, Landlord’s repair obligations with respect to the Building or as may benefit the Premises shall be limited to maintaining in good order and repair the roof, roof membrane and exterior surfaces of the exterior walls of the Building (exclusive of doors, door frames, door checks, other entrances, internal windows and window frames which are not a part of the Common Area), and in load bearing walls and columns and foundations and floor slabs which form a part of, or are contiguous to, the Premises and in the sanitary sewer and electrical and gas systems to their point of entry into and exit from the Premises, the sprinkler and fire alarm systems serving the Premises, and the Common Area (including keeping the Common Area and sidewalks free of snow and ice and lit at night); provided, however, that Landlord shall not be required to make any such repairs occasioned by the act or omission of Tenant, its agents, employees, licensees or contractors. Landlord shall not be required to make any improvements or repairs of any kind upon the Premises and appurtenances unless otherwise expressly provided in this Lease. Except if required due the negligence of Tenant, its agents, contractors and employees, Landlord shall be responsible for maintenance and repair of the HVAC System, and for full replacement should it be necessary, at Landlord’s sole cost and expense. Tenant shall promptly report in writing to Landlord any defective condition which Landlord is required to repair, and Landlord’s obligation to repair as set forth in this Section 11.01 is conditioned upon receipt by Landlord of such written notice. Landlord’s obligation to repair as set forth in this Section 11.01 is also conditioned upon Tenant not then being in default under this Lease.
Section 11.02. Tenant’s Obligations. Except as provided in Section 11.01, Tenant, at Tenant’s sole cost and expense, shall keep and maintain in (first class) good and safe order, condition and repair the Premises and every part thereof, and any and all appurtenances thereto wherever located, including, without limitation, the interior surfaces of the exterior walls, the exterior and interior portion of all doors, door frames, door checks, other entrances, interior windows, window frames and plate glass (for the avoidance of doubt, all exterior windows, window frames and plate glass shall not be considered a part of the Premises and as such shall not be the responsibility of the Tenant to repair and maintain, except if damaged as a result of the actions of Tenant or anyone claiming by, through or under Tenant), all plumbing and sewage facilities located entirely within and exclusively serving the Premises (unless the need for repair arises as a result of activities outside the Premises), wall coverings, floor coverings, ceilings and Tenant’s improvements, and shall make all other interior repairs, replacements, renewals and restorations, ordinary and extraordinary, foreseen and unforeseen, required to be made in and to the Premises, except as expressly set forth herein. The term “repair” as used in this Article 11 shall include replacements and/or renovations, when necessary. If Landlord shall have the benefit of any warranties from contractors relating to any of Tenant’s maintenance or repair responsibilities under this Section, Landlord shall, upon request, assign such warranties to Tenant (to the extent such warranties are assignable) or endeavor to enforce such warranties for Tenant’s benefit.
Section 11.03. Landlord’s Performance of Tenant’s Obligations. If Tenant fails to promptly perform its obligations as set forth in Section 11.02 within ten (10) days of written notice from Landlord, or in the case of an emergency, Landlord may (but shall not be obligated to) perform or cause to be performed such repairs without incurring any liability to Tenant for any damage caused thereby (except to the extent caused by the negligence or willful misconduct of Landlord or Landlord’s agents, employees or contractors), and Tenant shall pay to Landlord within thirty (30) days of Landlord’s demand (including reasonable backup documentation for such expenditures), as Additional Rent, the reasonable costs incurred by Landlord thereby plus interest thereon at the rate per annum of five percent (5%) above the prime rate of interest announced from time to time by Citibank, N.A. (or any successor thereto, or if there shall be no such successor, such other bank or financial institution as Landlord may designate in writing to Tenant) (the “Default Rate”) from the date incurred until the date paid.
Section 11.04. Surrender of Possession. Tenant shall surrender the Premises at the expiration of the Term broom clean and in as good condition as on the Commencement Date, or in such better
Page 19 of 60
condition as the Premises may be put during the Term, excepting only deterioration caused by ordinary wear and tear and damage resulting from fire and other casualty. On or prior to the expiration of the Term, Tenant shall remove all of its fixtures, furnishings, equipment, cabling, wiring, other items of personal property and all Special Alterations from the Premises (excluding Landlord’s Work as set forth in Article 6, which shall in no event be considered a Specialty Alteration) not permitted to remain by Landlord and repair any damage resulting therefrom. “Special Alterations” shall mean (i) raised flooring, (ii) alterations which would penetrate floor slabs or affect the exterior of the Building, (iii) safes, vaults, high density document storage systems (as opposed to typical office filing cabinets), or any other installation that would be more difficult or expensive to remove than typical Building standard office fit up, (iv) security systems(s), and (v) other alterations, additions or improvements reasonably identified by Landlord as inconsistent with those typically installed in the Building by tenants of similar space in the Building or in similar space in buildings of the same type and quality as the Building in the business district of Bridgewater, New Jersey.
Section 11.05. Tenant’s Performance of Landlord’s Obligations. Notwithstanding anything to the contrary in this Article 11, if Landlord fails to perform its maintenance obligation under this Lease with respect to the Premises which it is obligated to perform under this Article 11 within a reasonable period of time not to exceed seventy-five (75) (except in the case of an emergency threatening imminent harm to life or material damage to property) days following receipt of written notice from Tenant as set forth above, Tenant shall be permitted to perform such obligations in the Premises on Landlord’s behalf, provided Tenant first delivers to Landlord an additional three (3) business days prior written notice indicating that Tenant will be performing such obligations and provided Landlord fails to commence to perform its obligation(s) within such additional three (3) business day period or thereafter fails to diligently complete performance of such obligations having commenced performance within such three (3) business day period. If the obligations to be performed by Tenant will affect the Building’s Operating Systems, Tenant shall use only those contractors used by Landlord in the Building for work on such Operating Systems. All other contractors shall be subject to Landlord’s reasonable approval and Landlord agrees to approve or reject any contractor proposed to be used by Tenant within seventy-two (72) hours of receipt of Tenant’s second notice (unless Landlord has either commenced to perform its obligation(s) within such additional three (3) business day period or thereafter failed to diligently complete performance of such obligations having commenced performance within such three (3) business day period), provided that if a proposed contractor is duly licensed and bonded and all requisite permits have been obtained for the desired work, Landlord agrees not to withhold its approval of the proposed contractor. Landlord agrees to reimburse Tenant within thirty (30) days following receipt from Tenant of a written statement of all reasonable and actual out of pocket costs incurred by Tenant in performing such obligations on behalf of Landlord. Nothing contained in this paragraph shall be interpreted to mean that Tenant shall be excused from paying any Rental due in the event of any alleged default by Landlord. Notwithstanding anything to the contrary herein, under no circumstances shall Tenant have the right to perform any structural repairs that are Landlord’s obligation to perform under this Article 11 (except in the case of an emergency threatening imminent harm to life or material damage to property).
ARTICLE 12
ALTERATIONS, ADDITIONS AND IMPROVEMENTS
Section 12.01. Tenant’s Changes. The term “Tenant’s Changes” shall mean any and all alterations, installations, additions or improvements made or to be made by or on behalf of Tenant in or to the Premises. The term “Structural or Exterior Changes” as used in this Lease shall mean any and all alterations, installations, additions or improvements in or to the Premises which (i) affect the exterior of the Premises or are visible from outside the Premises at street level, or (ii) affect the structure of the Premises or any of its outer walls, any of its inner walls or columns which are load bearing, its foundation or roof, or (iii) affect any of the building or service systems located in the Premises or Building,
Page 20 of 60
including, without limitation, the mechanical, electrical, HVAC, plumbing, sprinkler and other service systems.
Section 12.02. Requirements. Tenant shall not make any Tenant’s Changes which shall have a cost in excess of $25,000 in any one instance or which shall constitute Structural or Exterior Changes without Landlord’s prior written consent, which consent may be withheld in Landlord’s sole and uncontrolled discretion as to any Structural or Exterior Change, and which consent shall not be unreasonably withheld, conditioned or delayed in relation to approval for other Tenant’s Changes. With respect to any Tenant’s Change, Tenant shall, at least ten (10) days prior to commencing any such change, deliver to Landlord a statement describing with precision and in detail the proposed change. In addition, for any Tenant’s Change requiring Landlord’s consent as set forth in the first sentence of this Section 12.02, Tenant shall deliver drawings and specifications therefor to Landlord for its written approval prior to commencing any such change. All Tenant’s Changes shall be performed in accordance with the following terms and conditions:
|
(i) Tenant shall obtain all necessary permits and approvals required under applicable Laws for the performance of Tenant’s Changes; |
|
(ii) Tenant shall perform Tenant’s Changes in a good and workmanlike manner and, if such changes require Landlord’s consent, strictly in accordance with the drawings and specifications previously approved by Landlord; |
|
(iii) Tenant shall perform Tenant’s Changes which are Structural Changes and Exterior Changes only by using contractors approved in writing by Landlord, provided, however that at Landlord’s option, Landlord shall have the right to perform any Structural or Exterior Changes on Tenant’s behalf with contractors of Landlord’s choosing, provided that the costs of such contractors shall be competitive with the costs of contractors performing similar work in similar buildings in Bridgewater, New Jersey; |
|
(iv) Tenant shall, at its sole cost and expense, carry, or cause to be carried, (i) worker’s compensation insurance in statutory limits covering all persons employed in connection with Tenant’s Changes, (ii) personal injury liability and property damage insurance in the aggregate sum of Five Million Dollars ($5,000,000) per occurrence, (iii) Builder’s risk insurance, completed value form with replacement cost endorsement, in an “agreed amount” sufficient to avoid any coinsurance; and (iv) such other insurance and in such amounts, as Landlord shall deem reasonably necessary; |
|
(v) Tenant shall obtain waivers of lien from all contractors, laborers and materialmen and shall discharge or bond, in accordance with the provisions of Section 12.04, any liens filed against the Premises or the Building; |
|
(vi) Tenant shall perform Tenant’s Changes in such a manner as shall not interfere with the construction, use or enjoyment of the remainder of the Building; |
|
(vii) Tenant shall pay to Landlord, within ten (10) days of request therefor, the actual, reasonable, third party out-of-pocket costs incurred by Landlord in connection with Landlord’s review of Tenant’s drawings and specifications for Tenant’s Changes; no review or approval by Landlord of Tenant’s drawings and specifications shall constitute any representation or warranty by Landlord as to the adequacy, correctness, efficiency, compliance with Laws or any other aspect of such drawings and specifications; and |
Page 21 of 60
|
(ix)If in connection with Tenant’s Changes, Tenant shall need to make any roof penetrations whatsoever to the Premises, Tenant shall utilize Landlord’s roofing contractor (which Landlord hereby advises Tenant is necessary in order to preserve Landlord’s roof warranty). |
Section 12.03. Removal. All Tenant’s Changes shall be deemed to have attached to the leasehold and to have become the property of Landlord upon such attachment. Upon the termination of this Lease, subject to Section 11.04, Tenant shall not remove any of such Tenant’s Changes (other than Tenant’s wiring and cabling) except that Tenant shall have the right to remove (and shall be obligated to remove, upon Landlord’s request) trade fixtures, furnishings and equipment installed by Tenant. Tenant shall repair any damage to the Premises and the Building caused by such removal.
Section 12.04. Liens. Tenant shall pay promptly all persons furnishing labor or materials in connection with Tenant’s Changes. Tenant shall not suffer or permit any liens to be filed against the Premises or the Building or any portion thereof or against Tenant’s leasehold estate therein, by reason of any work, labor, material or services done for, or supplied to or claimed to have been done for or supplied to Tenant or anyone claiming by, through or under Tenant. If any such lien shall be filed, Tenant shall, within twenty (20) days after written notice from Landlord of the filing thereof, either cause such lien to be vacated and canceled of record or, if Tenant in good faith determines that such lien should be contested, furnish such security, by surety bond or otherwise, as may be necessary or prescribed by Laws to release the lien and prevent any foreclosure of such lien during the pendency of such contest. If Tenant shall fail to vacate or cause the release of any lien within twenty (20) days after written notice from Landlord of the filing thereof, in addition to any other right or remedy of Landlord resulting from Tenant’s default, Landlord may, but shall not be obligated to, vacate or release such lien either by paying the amount claimed to be due or by giving security or in such other manner as may be prescribed by Laws. Tenant shall pay to Landlord, within thirty (30) days of Landlord’s written demand, all reasonable sums incurred by Landlord in connection therewith, including, without limitation, Landlord’s costs, expenses and attorneys’ fees, together with interest thereon at the Default Rate from the date incurred until the date paid.
ARTICLE 13
SIGNS
Section 13.01. Sign. Landlord shall, at its expense, place identification for Tenant in any present or future building monuments or directories, if any, and suite entrance(s). Tenant shall, at its expense, have the right to place its name or logo on the building exterior in not more than two (2) location(s), subject to both: (i) Landlords approval, not to be unreasonably withheld; and (ii) local code and any municipal authority requiring approval. In the event Landlord provides for signage in the lobby of the Building, Tenant shall be entitled to its share of such signage, consistent with signage granted to other tenants within the Building.
ARTICLE 14
INSURANCE AND INDEMNITY
Section 14.01. Tenant’s Insurance. Tenant, at its sole cost and expense, shall obtain and maintain in full force and effect during the Term the following insurance coverages with respect to the Premises:
Page 22 of 60
(i)Commercial general liability insurance, with contractual liability endorsement, with a combined single limit per occurrence for personal injury and property damage of not less than Three Million Dollars ($3,000,000.00), with a Four Million Dollar ($4,000,000.00) aggregate limitation;
(ii)“Special Form” (formerly known as “all-risk”) property insurance insuring loss of or damage to the Premises and all Tenant's personal property located therein, including, without limitation, Tenant's goods, trade fixtures, and equipment, written at 100% of replacement cost (exclusive of footings, foundations and underground utilities) with an “agreed amount” sufficient to avoid coinsurance;
(iii) Builder's Risk insurance as set forth in Article 12;
(iv) Worker's Compensation and Employer’s Liability insurance as required by Laws; and
(v) Such other insurance as reasonably requested by Landlord from time to time.
Section 14.02. Form. Each insurance policy shall be written in the name of Tenant, and shall name (except the worker’s compensation policy) Landlord and any other parties in interest (including mortgagees) designated by Landlord as additional insureds. Each policy shall be written by a nationally recognized insurance company with a Best’s Rating of A VIII or better, or an equivalent rating by a similar or successor authority, and legally licensed to do business in the State of New Jersey. The coverage limits of each policy shall be increased from time to time as reasonably required by Landlord.
Section 14.03. Special Clauses. Each such policy shall contain the following endorsements, provisions and/or clauses: (i) a provision that such policy and the coverage evidenced thereby shall be primary and non-contributing with respect to any policies carried by Landlord, and that any coverage carried by Landlord shall be excess insurance (this provision shall be applicable to Tenant’s property insurance policy only); and (ii) a provision that the insurer will not cancel, change in any material or adverse respect or fail to renew the coverage provided by such policy without first giving Landlord and all additional insureds at least thirty (30) days’ prior written notice; provided, however, in the event Tenant’s insurance provider shall not provide such notice, Tenant shall be obligated to provide Landlord with the same.
Section 14.04. Delivery of Policies. Tenant shall deliver to Landlord on or before the earlier to occur of (i) the Commencement Date, and (ii) the date the Tenant or Tenant’s contractor’s enter the Premises to perform Tenant’s Work, and thereafter at least fifteen (15) days prior to the expiration of each policy, an insurance certificate evidencing each policy required under this Lease to be procured by Tenant, together with evidence satisfactory to Landlord of full payment of the premiums therefor.
Section 14.05. Landlord’s Right to Obtain Insurance on Tenant’s Behalf. If Tenant fails to procure, maintain and/or pay for, at the times and for the durations specified in this Article 14, any insurance required under this Lease, or fails to carry any other insurance required by Laws, if such failure is not cured by Tenant within ten (10) days of written notice thereof by Landlord, Landlord may (but without obligation to do so) at any time or from time to time, and without further notice, procure such insurance and pay the premiums therefor on Tenant’s behalf, in which event Tenant shall pay to Landlord, as Additional Rent, all reasonable sums so paid by Landlord and any reasonable costs or expenses incurred by Landlord in connection therewith, together with interest thereon at the Default Rate from the date incurred until the date paid.
Page 23 of 60
Section 14.06. Separate Insurance. Tenant shall not carry any separate insurance concurrent in form or contributing in the event of loss with that required in this Article 14 unless Landlord is included therein as an additional insured.
Section 14.07. Increase in Landlord’s Insurance Premium. Tenant shall not use the Premises in a manner which will in any way impair or invalidate any policy of insurance covering the Premises or the Building, provided the same is not intended to prohibit Tenant from using the Premises for the Permitted Use. Tenant shall pay within thirty (30) days of Landlord’s written demand, as Additional Rent, any increase in premiums for insurance Landlord may reasonably elect to carry on the Premises or the Building, resulting from Tenant’s manner of use of the Premises, beyond use as general, executive and administrative office and training space.
Section 14.08. Non-Liability of Landlord. Except as to gross negligence or willful misconduct, neither Landlord nor its agents shall be liable to Tenant for any loss, injury or damage to Tenant or to any other person, or to its or their property, irrespective of the cause of such injury, damage or loss, nor shall the aforesaid parties be liable for any damage to property of Tenant or of others entrusted to employees of Landlord nor for loss of or damage to any such property by theft or any other reason whatsoever, including, without limitation, damage caused by or resulting from bursting, stoppage, leaking or freezing of water, gas, sewer or steam pipes. Further, neither Landlord nor its agents, even if negligent, shall be liable for consequential damages arising out of any loss of use of the Premises or any equipment, facilities or other Tenant’s property therein by Tenant or any person claiming through or under Tenant.
Section 14.09. Indemnification. Tenant shall indemnify, hold harmless and defend Landlord, its officers, directors, stockholders, beneficiaries, partners, representatives, agents and employees, from and against any and all Liabilities arising from or relating to: (i) any occurrence in, upon or at the Premises unless caused by the negligence or willful misconduct of Landlord, its agents, employees and contractors, (ii) the occupancy or use of the Premises or any part thereof by Tenant, its contractors, agents, employees, subtenants, licensees, invitees and concessionaires, (iii) a breach of the provisions of this Lease by Tenant, its subtenants, licensees and concessionaires and their agents, contractors, employees, invitees or licensees or (iv) any wrongful act of omission of Tenant, its subtenants, licensees and concessionaires and their agents, contractors, employees, invitees or licensees. If Landlord or any other party so indemnified shall, without fault, be made a party to any litigation commenced by or against Tenant, or if Landlord or any such party shall, in its sole discretion, determine that it must intervene in such litigation to protect its interests, then Tenant shall pay all costs, expenses and reasonable attorneys’ fees incurred or paid by such party in connection with such litigation.
Landlord shall indemnify, defend, and hold harmless Tenant and its agents, employees, contractors and invitees from and against any and all claims, actions, damages, liability and expense which may be asserted against, imposed upon, or incurred by Tenant or its agents, employees, contractors and invitees and (i) arising out of or in connection with loss of life, personal injury or damage to property to the arising from or related to any occurrence in, upon or at the Common Area, or (ii) resulting from the gross negligence or willful misconduct of Landlord or any of Landlord’s employees, agents or contractors, all unless caused by the negligence or willful misconduct of Tenant, its agents, employees and contractors.
Section 14.10. Waiver of Direct Action and Subrogation. Landlord and Tenant hereby release each other from any and all liability or responsibility to the other or anyone claiming through or under them by way of subrogation or otherwise for any loss or damage specifically insured against or required by the terms hereof to be insured or self-insured against by such party, even if such loss or damage shall have been caused by the fault or negligence of the other party, or anyone for whom such party may be responsible, except to the extent of a standard deductible under such policy.
Page 24 of 60
Section 14.11. Survival. This Article 14 shall survive the expiration or earlier termination of this Lease.
Section 14.12. Landlord’s Insurance Requirements. Landlord shall maintain (i) insurance against loss or damage to the Building or the Property with coverage for perils as set forth under the “Causes of Loss-Special Form” or equivalent property insurance policy in an amount equal to the full insurable replacement cost of the Building (excluding coverage of Tenant’s personal property and any Tenant Changes), (ii) commercial general liability insurance, including premises liability and contractual liability insurance and (iii) rent loss coverage sufficient to cover one year of rent for the Building. The commercial general liability policy shall insure against claims for bodily injury, personal injury, death or property damage occurring on, in or about the Property with limits of not less than $1,000,000.00 per occurrence with umbrella policy coverage of $5,000,000.00. The deductible for any insurance policy required hereunder must not exceed $10,000.00.
ARTICLE 15
DAMAGE BY FIRE OR OTHER CASUALTY
Section 15.01. Landlord’s Restoration. If the Premises, or access thereto, shall be partially or totally damaged or destroyed by fire or other casualty (“Casualty”), Tenant shall immediately notify Landlord of the details of such damage or destruction. If more than twenty-five percent (25%) of the rentable area of the Premises, or access thereto, has been damaged or destroyed during the last twelve (12) months of the Term, then either Landlord or Tenant may terminate this Lease by giving ten (10) days’ notice thereof to the other party, such notice to be given within sixty (60) days of the date of the Casualty. If (i) more than twenty-five percent (25%) of the gross leasable area of the building in which the Premises is located or more than twenty percent (20%) of the rentable area of the Building shall be damaged or destroyed, or (ii) if any damage caused by a Casualty by the occurrence of any risk not fully insured under Landlord’s property damage insurance policy, then Landlord may terminate this Lease upon giving ten (10) days’ notice thereof to Tenant, such notice to be given within one hundred fifty (150) days of the date of the Casualty. If this Lease is not terminated in accordance with the foregoing, Landlord shall promptly repair and restore the exterior walls and the roof and structural support columns of the Premises to substantially the condition existing on the date the Premises were first delivered to Tenant, provided, however, that Landlord shall be obligated to perform such restoration only to the extent of the net insurance proceeds therefor paid to Landlord under Landlord’s property damage insurance policy, and Tenant shall, at Tenant’s sole cost and expense, repair and restore Tenant’s Work (including Tenant’s trade fixtures), and Tenant’s merchandise, furnishings and equipment and reopen for business.
Section 15.02. Abatement of Rent. If the Premises shall be partially or totally damaged by a Casualty (not caused by the gross negligence or willful misconduct of Tenant or any of its agents or employees), Base Rent and all Additional Rent shall be abated in proportion to the rentable area of the Premises rendered untenantable by the Casualty and thereafter actually not used by Tenant for the conduct of its business, such abatement to commence on the date of the Casualty and to continue until fifteen (15) days after the date of substantial completion of Landlord’s restoration work.
ARTICLE 16
EMINENT DOMAIN
Section 16.01. Total Condemnation. If any portion of the Premises shall be taken by any public authority under the power of eminent domain or sold to public authority under threat of or in lieu of such a taking (any or all of the foregoing shall constitute a “Taking” and the property subject to such action shall be deemed to have been “Taken”), then the Term shall cease as the date possession shall be taken by such public authority, and the Rental shall be paid up to such date, with a proportionate refund
Page 25 of 60
by Landlord to Tenant of such Rental as may have been paid in advance for a period subsequent to the date of the Taking.
Section 16.02. Partial Condemnation - Landlord’s Termination Right. If less than the whole of the Property shall be Taken, the Landlord may, upon giving written notice thereof to Tenant on or before the thirtieth (30) day following the date possession is surrendered to the public authority, terminate this Lease as of the day possession is taken by the public authority. The Rental and other charges shall be paid up to the date possession is taken by the public authority, with an appropriate refund by Landlord to Tenant of such Rental as may have been paid in advance for a period subsequent to the date of the Taking.
Section 16.03. Landlord’s and Tenant’s Damages. All damages awarded for a Taking, whether for the whole of or a part of the Premises, shall belong to and be the property of Landlord. Nothing contained herein, however, shall prevent Tenant from pursuing a separate claim against the condemning authority for the value of furnishings, equipment and trade fixtures installed in the Premises at Tenant’s expense and for relocation expenses, provided that such claim shall in no way diminish the award or compensation payable to or recoverable by Landlord in connection with such taking or condemnation.
ARTICLE 17
ASSIGNMENT AND SUBLETTING
Section 17.01. Assignment or Subletting. Except as expressly set forth herein, Tenant shall not assign or in any manner transfer, mortgage or encumber this Lease or any estate or interest therein, nor lease or sublet the Premises or any part thereof or any right or privilege appurtenant thereto, nor allow anyone to conduct business at, upon or from the Premises (whether as a concessionaire, franchisee, licensee, permittee, subtenant, department operator or otherwise) or to come in, by, through or under it, in all cases either by the voluntary or involuntary act of Tenant or by operation of law or otherwise. Except as set forth in Section 17.09 below and in the case of sales or transfers among existing shareholders of Tenant on the date hereof or redemption of stock by the Tenant corporation, the sale, issuance or transfer of any voting capital stock of Tenant, or of any corporate entity which directly controls Tenant (if Tenant or such controlling corporate entity is a corporation the stock of which is not publicly traded), or any interest in any non-corporate entity which directly controls Tenant, which results in a change in the direct voting control of Tenant, shall be deemed to be a prohibited assignment of this Lease within the meaning of this Section 17.01. Except as set forth in Section 17.09 below and if Tenant is a partnership, trust or unincorporated association, then the sale, issuance or transfer of a controlling interest therein, or the transfer of a majority interest in or a change in the voting control of any partnership, trust, unincorporated association, or corporation which directly controls Tenant, or the transfer of any portion of any general partnership or managing interest in Tenant, shall be deemed to be a prohibited assignment of this Lease within the meaning of this Section 17.01. Any purported assignment, subletting, transfer or other act prohibited under this Section 17.01 shall be null and void and constitute a default under this Lease.
Section 17.02. Tenant’s Request to Assign or Sublet. If Tenant shall desire to assign or sublet this Lease, Tenant shall, at least thirty (30) days prior to the effective date of any proposed assignment or sublease, by notice given as provided in Section 32.09 (“Tenant’s Notice”), furnish Landlord with (i) the name and address of the proposed assignee; (ii) the terms, conditions and consideration of the proposed assignment; (iii) current financial information with respect to the proposed assignee, including, without limitation, a current financial report; and (iv) any other information as Landlord may reasonably request with respect to the proposed assignee. Landlord shall provide Tenant with its consent or disapproval of any such request for an assignment or sublet within thirty (30) days following receipt of Tenant’s Notice.
Page 26 of 60
Section 17.03. Intentionally Omitted.
Section 17.04. Landlord’s Consent. Upon Tenant’s compliance with the provisions of Section 17.02, Landlord’s consent shall not unreasonably be withheld, conditioned or delayed to the proposed assignment or subletting, provided and upon condition that:
(i) the assignee or subtenant shall have a good business reputation;
(ii) the assignee or subtenant proposes to use the Premises for the Permitted Use;
(iii)the proposed assignee or sublessee is not an entity who is then a tenant in the Building or an entity with which Landlord is then negotiating or within four (4) months has negotiated for space in the Building, unless at the time such request is made to Landlord, Landlord does not have available space in the Building for the proposed assignee or sublessee;
(iv) Tenant shall not be in default under this Lease, beyond applicable notice and cure periods, either at the time Landlord’s consent to such assignment is requested or on the effective date of the proposed assignment or subletting;
(v)there shall be delivered to Landlord evidence for the assignee or the subtenant of the same insurance coverages as are required to be carried by the Tenant pursuant to Section 14.01; and
(vi)if a subletting, the subletting is made subject to all of the obligations of Tenant under this Lease and, without limiting the generality of the foregoing, the sublease specifically provides that there shall be no further subletting of the sublet premises or an assignment thereof other than in strict accordance with the terms of this Article 17.
Section 17.05. Approved Sublettings. Tenant shall furnish Landlord with a copy of an executed counterpart of each sublease within ten (10) days after the date of its execution. No sublease shall be valid and no subtenant shall take possession of the Premises or any part thereof until such executed counterpart has been delivered to Landlord. The form of sublease shall be reasonably acceptable to Landlord and shall provide for a sublease term ending not later than one (1) day prior to the expiration date of the Term. Such sublease shall further provide that it is subject and subordinate to this Lease and to the matters to which this Lease is or shall be subordinate, and that in the event of a termination, re-entry or dispossession by Landlord under this Lease, Landlord may, at its option, succeed to all of the right, title and interest of Tenant, as sublessor under such sublease, and such subtenant shall, at Landlord’s option, attorn to Landlord pursuant to the then executory provisions of such sublease, provided, however, that Landlord shall not (i) be liable for any previous act or omission of Tenant under such sublease, (ii) be subject to any offset not expressly set forth in such sublease which theretofore accrued to such subtenant against Tenant, or (iii) be bound by any previous prepayment of more than one month’s fixed rent under such sublease.
Section 17.06. Approved Assignments. Tenant shall furnish to Landlord a counterpart of each assignment within ten (10) days of the date of its execution. No assignment shall be binding upon Landlord and no assignee shall take possession of the Premises or any part thereof (including, without limitation, a permitted assignee under Section 17.09 hereof) unless Tenant shall, concurrent with the delivery of an executed counterpart of such assignment, deliver to Landlord an agreement executed by the assignee, in appropriate form for recording, whereby such assignee agrees unconditionally to be bound by and to perform all of the obligations of Tenant under this Lease arising after the date of the assignment and further agrees that notwithstanding such assignment, the provisions of this Article 17 shall continue to be binding upon such assignee with respect to all future assignments and transfers.
Page 27 of 60
Section 17.07. Landlord’s Costs. Tenant shall pay, as Additional Rent, Landlord’s reasonable third party out-of-pocket costs incurred in connection with any subletting or assignment permitted or proposed by Tenant, whether or not consented to by Landlord, including, without limitation, reasonable attorneys’ fees and the costs of credit checks and reports, in an amount not to exceed $5,000 per request. Such Additional Rent shall be payable by Tenant within ten (10) days after Landlord’s written demand therefor.
Section 17.08. Excess Consideration. Except in the event of an assignment to an entity purchasing substantially all of the assets of Tenant's business or any Permitted Transfer in accordance with Section 17.09 below, in the event of any assignment or subletting under this Lease, Tenant shall pay to Landlord, as Additional Rent:
|
(i)in the case of an assignment, an amount equal to fifty percent (50%) of all sums paid to Tenant by the assignee for or by reason of such assignment, including, without limitation, sums paid for the sale of Tenant's Changes (including Tenant's trade fixtures) and Tenant's merchandise, furnishings and equipment, less the then net unamortized or undepreciated cost thereof determined on the basis of Tenant's federal income tax returns and less Tenant's other reasonable costs actually incurred in connection with the assignment, including reasonable brokerage and attorneys' fees; and |
|
(ii)in the case of a sublease, an amount equal to fifty percent (50%) of all sums payable under the sublease to Tenant by the subtenant which exceed the Base Rent and Additional Rent accruing under this Lease during the term of the sublease in respect of the subleased space (at the rate per square foot of gross leasable area payable by Tenant under this Lease), including, without limitation, sums paid for the sale or rental of Tenant's Changes (including Tenant's trade fixtures) and Tenant's merchandise, furnishings and equipment, (less, in the case of the sale thereof, the then net unamortized or undepreciated cost thereof determined on the basis of Tenant's federal income tax returns) and less Tenant's other reasonable costs actually incurred in connection with the subletting, including reasonable brokerage and attorneys' fees, rent concessions and costs of outfitting the subleased premises for the subtenant. |
The sums payable under this Section 17.08 shall be paid to Landlord as and when payable by the assignee or subtenant as the case may be, to Tenant.
Section 17.09. Permitted Transfer. Notwithstanding anything to the contrary set forth in this Lease, provided and upon condition that Tenant shall comply with the provisions of Sections 17.02 and 17.06 hereof, on fifteen (15) days’ prior written notice to Landlord, Tenant may assign or sublet this Lease without Landlord’s consent to (i) to an Affiliate (as hereinafter defined) of Tenant or (ii) in connection with a merger or consolidation of Tenant or (iii) in the case of a sale of substantially all of the assets or corporate stock of Tenant, provided that in the case of (ii) or (ii) above, the assignee or subtenant shall have a liquid net worth consisting of cash, cash equivalents and accounts receivable no older than ninety (90) days (less customary reserves for bad debts) of not less than Two Hundred Million Dollars ($200,000,000.00) (“Permitted Transfer”). Tenant shall furnish to Landlord a counterpart of any Permitted Transfer within thirty (30) days of the date of its execution and any proof reasonably requested by Landlord substantiating the such assignment or sublet is a Permitted Transfer (including financial statements certified by an independent public accountant, if applicable). No Permitted Transfer shall be binding upon Landlord unless Tenant shall, concurrent with the delivery of an executed counterpart of such Permitted Transfer, deliver to Landlord, in accordance with this provision, an agreement executed by the assignee or subtenant whereby such assignee or subtenant agrees unconditionally to be bound by and to perform all of the obligations of Tenant under this Lease arising after the date of the assignment or sublet, as may be applicable, and further agrees that notwithstanding such assignment or sublet, the provisions of this Article 17 shall continue to be binding upon such assignee or subtenant with respect to all
Page 28 of 60
future assignments and transfers. The term “Affiliate” as used in this Section 17.09 shall mean any entity that controls, is controlled by or is under common control with Tenant. Any subsequent transfer by an Affiliate or other entity to whom a transfer is permitted under this Section 17.09 shall again be subject to all of the terms and conditions of this Lease. “Control,” as used in this Article 17, shall mean the ownership, directly or indirectly, more than fifty percent (50%) of the voting securities of, or possession of the right to vote, in the ordinary direction of its affairs, of more than fifty percent (50%) of the voting interest in, any person or entity. Furthermore, notwithstanding anything herein to the contrary, employees of an Affiliate of Tenant shall be permitted to occupy and use the Premises pursuant to the terms of this Lease and such occupancy shall not be deemed a sublease or assignment in violation of the terms of this Lease so long as such Affiliate is not paying rent for such occupancy.
Section 17.10. Tenant Remains Liable. Notwithstanding any assignment of this Lease, Tenant shall remain fully liable for the payment of the Rental due and to become due under this Lease and the terms, provisions, and conditions contained in this Lease on the part of Tenant to be performed. The consent by Landlord to an assignment shall not in any way be construed to relieve Tenant from its obligation to obtain the consent in writing of Landlord to any further assignment. If Tenant assigns, transfers, mortgages or encumbers this Lease or any interest therein or sublets all or any portion of the Premises in violation of the provisions of this Article 17, or if the Premises are occupied by anyone other than Tenant, Landlord may collect rent from any such assignee, sublessee or anyone who claims a right to this Lease or who occupies the Premises, and Landlord may apply the net amount collected to the Rental, and no such collection shall be deemed a waiver by Landlord of any of the terms, provisions, and conditions contained in this Article 17 nor an acceptance by Landlord of any such assignee, sublessee, claimant or occupant as Tenant, nor be deemed to release Tenant from the further performance of all of Tenant’s obligations under this Lease. If Landlord shall decline to give its consent to any proposed assignment, Tenant shall indemnify and hold harmless and defend Landlord from and against any Liabilities arising from or relating to any claims that may be made against Landlord by the proposed assignee or by any brokers or other persons claiming a commission or similar compensation in connection with the proposed assignment.
ARTICLE 18
CONDITIONS OF LIMITATION
Section 18.01. Bankruptcy or Insolvency of Tenant. To the extent permitted by Laws, this Lease, and the Term and estate hereby granted, are subject to the limitation that, whenever Tenant shall make an assignment for the benefit of creditors, or shall consent to, or acquiesce in, the appointment of a liquidator, receiver, trustee, or other custodian for itself or for the whole or any part of its properties or assets, or shall commence a voluntary case for relief under the United States Bankruptcy Code (“Bankruptcy Code”) or file a petition or take advantage of any bankruptcy or insolvency act or applicable law of like import, or whenever an involuntary case under the Bankruptcy Code shall be commenced against Tenant, then, Landlord (a) at any time after Landlord learns of any such event, or (b) if such event occurs without the acquiescence of Tenant, at any time after the event continues for sixty (60) days, may give Tenant a notice of intention to end the Term upon the expiration of three (3) days from the date of service of such notice of intention, and upon the expiration of such three (3) day period, this Lease and the Term and estate hereby granted shall terminate with the same effect as if such date were the Expiration Date, provided, however, that Tenant shall remain liable for damages as provided in Article 20.
Section 18.02. Rights and Obligations Under the Bankruptcy Code. Upon the filing of a petition by or against Tenant under the Bankruptcy Code, Tenant, as debtor and/or as debtor in possession, and any trustee who may be appointed, agree to perform each and every obligation of Tenant under this Lease, including, without limitation, the manner of “operation” as provided in Article 7, until such time as this Lease is either rejected or assumed by order of the United States Bankruptcy Court. If
Page 29 of 60
this Lease is assumed, whether by Tenant or any trustee or by any assignee or successor to Tenant or such Trustee (the “Assuming Entity”), in addition to any other conditions or obligations imposed upon the Assuming Entity, shall be the following obligations and conditions: (i) the cure of any monetary defaults and the reimbursement of any pecuniary loss of Landlord, such cure to be made immediately upon entry of a court order providing for assumption by and/or assignment to the Assuming Entity; (ii) the deposit of a sum equal to two (2) months’ Base Rent and other charges, to be held pursuant to the terms of Article 27 of this Lease; (iii) the use of the Premises for the Permitted Use; (iv) the payment of Tenant’s Proportionate Share of any Operating Expenses which may then be due or which may thereafter become due pursuant to the provisions of Articles 8 and 9; (v) the Assuming Entity demonstrates in writing that it has sufficient background, including, without limitation, substantial experience and financial ability to operate an establishment out of the Premises in the manner contemplated in this Lease, as did Tenant at the time of the execution of this Lease; (vi) the prior consent has been obtained of any mortgagee or holder of a deed of trust to which this Lease has been assigned as collateral security, if necessary; and (vii) no physical changes of any kind may be made to the Premises unless in compliance with the applicable provisions of this Lease. Any person or entity to which this Lease is assigned pursuant to the provisions of the Bankruptcy Code shall be deemed without further act or deed to have assumed all of the obligations arising under this Lease on and after the date of such assignment. Any such assignee shall, upon demand, execute and deliver to Landlord an instrument confirming such assumption as Landlord shall reasonably request.
Section 18.03. Other Conditions of Limitation. This Lease, and the Term and estate hereby granted, are subject to the further limitations that:
(a) If Tenant shall fail to pay on the day same is due and payable any Rental and any such Rental shall remain unpaid for ten (10) days after Landlord shall have given a written notice to Tenant specifying Tenant’s failure to do so; provided, however, that Landlord shall not be required to give such notice more than two (2) times during any calendar year, then, thereafter, any such failure to pay Rental on the date when same is due and payable shall entitle Landlord to exercise the rights hereinafter provided in the event of Tenant’s default without further notice; or
(b)If Tenant shall do or permit anything to be done, whether by action or inaction, contrary to any of Tenant’s obligations or covenants under this Lease, and if such situation shall continue and shall not be remedied by Tenant within twenty (20) days after Landlord shall have given to Tenant a written notice specifying the same, or in the case of a happening or default which cannot with due diligence be cured within a period of twenty (20) days and the continuance of which for the period required for cure will not subject Landlord to the risk of criminal liability or termination of any superior lease or foreclosure of any superior mortgage or deed of trust, if Tenant shall not (i) within such twenty (20) day period advise Landlord of Tenant’s intention to duly institute all steps necessary to remedy such situation, (ii) duly institute within such twenty (20) day period, and thereafter diligently prosecute to completion all steps necessary to remedy the same, and (iii) complete such remedy within such time after the date of the giving of such notice to Landlord as shall reasonably be necessary;
Then, in any of the foregoing events, Landlord may give Tenant a notice of intention to end the Term at the expiration of three (3) days from the date of such notice and, upon the expiration of such three (3) day period, this Lease and the Term and estate hereby granted shall expire and terminate with the same effect as if such day were the Expiration Date, provided, however, that Tenant shall remain liable for damages as provided in Article 20.
Notwithstanding anything to the contrary contained in the Lease, Landlord shall in no event be in default in the performance of any of Landlord’s obligations under this Lease unless Landlord shall have failed to perform such obligations within thirty (30) days (or such additional time as is reasonably
Page 30 of 60
required to correct any such default, provided Landlord commences cure within 30 days) after notice by Tenant to Landlord properly specifying wherein Landlord has failed to perform any such obligation.
ARTICLE 19
RE-ENTRY BY LANDLORD
Section 19.01. Re-entry. If any of the events described in Section 18.01 or subparagraphs (a) or (b) of Section 18.03 shall occur, or upon the Expiration Date, whether or not Landlord has elected to terminate this Lease, Landlord or Landlord’s agents and employees may immediately or at any time thereafter re-enter the Premises or any part thereof, in the name of the whole, either by summary dispossess proceedings or by any suitable action or proceeding at law, without being liable to indictment, prosecution, or damages therefor, and may repossess the same, and may remove any persons therefrom, to the end that Landlord may have, hold and enjoy the Premises again as and of its first estate and interest therein. The word “re-enter” as used in this Article 19 is not restricted to its technical legal meaning. If this Lease is terminated under the provisions of Article 18, or if Landlord shall re-enter the Premises under the provisions of this Article 19 or in the event of the termination of this Lease or of re-entry, by or under any summary dispossess or other proceeding or action or any provision of Laws by reason of Tenant’s default under this Lease, beyond applicable notice and cure periods, Tenant shall pay to Landlord the Rental payable by Tenant to Landlord through the date of such termination, or through the date of such recovery of possession, as the case may be, and shall also pay to Landlord damages as provided in Article 20.
Section 19.02. Other Remedies. In the event of a breach by Tenant of any of its obligations under this Lease, Landlord shall also have the right of injunction. The special remedies to which Landlord may resort under this Lease are cumulative and are not intended to be exclusive of any other remedies or means of redress to which Landlord may lawfully be entitled at any time, and Landlord may invoke any remedy allowed at law or in equity as if specific remedies were not provided for in this Lease.
Section 19.03. Retention of Monies in Landlord’s Possession. If this Lease is terminated under the provisions of Article 18, or if Landlord shall re-enter the Premises under the provisions of this Article 19, or in the event of the termination of this Lease or of re-entry, by or under any summary dispossess or other proceeding or action or any provision of Laws by reason of Tenant’s default under this Lease, beyond applicable notice and cure periods, Landlord shall be entitled to retain all monies, if any, paid by Tenant to Landlord, whether as advance Rental, security, or otherwise, but such monies shall be credited by Landlord against any Rental due from Tenant at the time of such termination or re-entry or, at Landlord’s option, against any damages payable by Tenant under Article 20 or pursuant to Laws.
ARTICLE 20
DAMAGES
Section 20.01. Measure of Damages. If this Lease is terminated under the provisions of Article 18, or if Landlord shall re-enter the Premises under the provisions of Article 19, or in the event of the termination of this Lease or of re-entry, by or under any summary dispossess or other proceeding or action or any provision of Laws by reason of Tenant’s default under this Lease, beyond applicable notice and cure periods, Tenant shall pay to Landlord within fifteen (15) days of Landlord’s written demand as damages, in a single lump sum, the total of:
|
(a) all Base Rent and Additional Rent due and payable and unpaid under this Lease as of the date of Landlord’s reentry, termination and/or dispossession by summary proceedings or otherwise; |
Page 31 of 60
|
(c) all reasonable third party out of pocket costs and expenses incurred by Landlord in connection with its termination of this Lease and/or recovery of possession of the Premises and/or in removing all persons and property therefrom and/or recovering from Tenant the Rental and damages specified in this Article 20 or any other sums and damages to which Landlord may be entitled under applicable Laws; and |
|
(d) all reasonable costs and expenses incurred by Landlord in curing any covenant or condition on the part of Tenant to be observed or performed under this Lease which Tenant shall have failed to perform, following any applicable notice and cure periods, as of the date of such termination or reentry absent termination. |
In addition to the foregoing amounts, Tenant shall remain liable for and shall pay, on the days originally fixed under this Lease for the payment thereof, amounts equal to the installments of Rental reserved under this Lease as would, under the terms of this Lease, become due and payable if this Lease had not been terminated or Landlord had not reentered the Premises absent termination, whether the Premises be relet, or remain vacant in whole or in part for the remainder of the Term of for a period less than the remainder of the lease term, up to but not exceeding the amount of any deficiency then existing after giving due credit for any net proceeds of any reletting after deducting all of Landlord’s reasonable costs and expenses incurred in connection with such reletting of the Premises or any portion thereof for the whole or any part of the remainder of the then current Term or for a longer period (which reletting Landlord may do at its election, either in its name or as agent for Tenant), including, without limitation, brokerage and attorneys’ fees in connection with any new lease, and reasonable costs of Landlord in repairing or altering the Premises for the new tenant and any reasonable tenant allowance or other concessions granted or paid by Landlord. The failure of Landlord to re‑let the Premises or any part thereof shall not release Tenant or affect Tenant’s liability for damage. If the Premises or any part thereof should be relet in combination with other space, then proper apportionment on a square foot basis (for equivalent space) shall be made of the rent received from such reletting and of the expenses of reletting. If the Premises or any part thereof be relet by Landlord for the unexpired portion of the Term or any part thereof, then the amount of rent reserved upon such reletting shall, prima facie, be the fair and reasonable rental value for the Premises, or part thereof, so relet during the term of the reletting. Suit or suits for the recovery of such deficiency or damages, or for a sum equal to any installment or installments of Rental payable under this Lease, may be brought by Landlord from time to time at Landlord’s election, and nothing herein contained shall be deemed to require Landlord to await the date on which the lease term hereof would have expired by its own terms had there been no such default by Tenant or no such termination or reentry absent termination. In no event shall Tenant be entitled to receive any excess of such net rents over the Rental payable by Tenant to Landlord under this Lease, nor shall Tenant be entitled, in any suit for the collection of damages pursuant to this Article 20 to a credit in respect of any net rents from a reletting except to the extent that such net rents are actually received by Landlord.
Section 20.02. No Limitation of Remedies. Suit or suits for the recovery of such damages, or any installments thereof, may be brought by Landlord from time to time at its election, and nothing contained herein shall be deemed to require Landlord to postpone suit until the Expiration Date. Nothing herein contained shall be construed to limit or preclude recovery by Landlord of any sums or damages to which, in addition to the damages provided above, to which Landlord may lawfully be entitled by reason of Tenant’s default under this Lease, beyond applicable notice and cure periods. Nothing herein contained shall be construed to limit or prejudice the right of Landlord to prove for and obtain as liquidated damages by reason of the termination of this Lease or reentry into the Premises, an amount equal to the maximum allowed by any Laws in effect at the time when, and governing the proceedings in which, such damages are to be proved, whether or not such amount be greater, equal to, or less than any
Page 32 of 60
of the sums referred to in Section 20.01; provided; however, in no event shall Landlord be entitled to an acceleration of rents regardless of the availability of such a remedy under any applicable Laws.
ARTICLE 21
WAIVERS
Section 21.01. Waiver of Right of Redemption. Tenant hereby waives all right and privilege, under, or by reason of, any present or future Laws, to redeem the Premises or to have a continuance of this Lease for the Term after being dispossessed or ejected therefrom by process of law.
Section 21.02. Waiver of Right to Jury Trial. Tenant and Landlord hereby waive trial by jury in any action, proceeding, or counterclaim brought by Landlord against Tenant or Tenant against Landlord, as the case may be, with respect to any matter whatsoever in connection with this Lease, or the use or occupancy of the Premises.
Section 21.03. Waiver of Right to Counterclaim. Tenant hereby waives any right to interpose any noncompulsory counterclaim in any summary action or proceeding brought by Landlord in connection with this Lease.
ARTICLE 22
ESTOPPEL CERTIFICATES, ATTORNMENT AND SUBORDINATION
Section 22.01. Estoppel Certificates. Tenant shall, without charge, at any time and from time to time (but not more than once in any Twelve (12) month period except in the event of a sale or refinancing of the Building), within ten (10) days after receipt of written notice therefor from Landlord or from any mortgagee under any mortgage encumbering the Building, deliver, in recordable form, a duly executed and acknowledged certificate or statement to the party requesting such certificate or statement or to any other person, firm or corporation reasonably designated, certifying: (a) that this Lease is unmodified and in full force and effect, or, if there has been any modification, that this Lease is in full force and effect as modified, and stating any such modification; (b) the Commencement Date; (c) that the Rental is paid currently without any off-set or defense thereto, except as expressly set forth in the Lease; (d) the dates to which the Rental has been paid, and the amount of Rental, if any, paid in advance; (e) whether or not there is then existing any claim of Landlord’s default under this Lease and, if so, the nature thereof; and (f) any other matters relating to the status of this Lease as shall be reasonably requested, provided that, in fact, such facts are accurate and ascertainable.
Section 22.02. Attornment. If any proceedings are brought for the foreclosure of, or in the event of the conveyance by deed in lieu of foreclosure of, or in the event of exercise of the power of sale under, any mortgage made by Landlord covering the Premises, or if Landlord sells, conveys or otherwise transfers its interest in the Building or any portion thereof containing the Premises, or in the event the lessor under any superior lease shall succeed to Landlord’s interests under this Lease, this Lease shall remain in full force and effect and Tenant hereby attorns to, and covenants and agrees to execute an instrument in writing reasonably satisfactory to the new owner and Tenant whereby Tenant attorns to such successor in interest and recognizes such successor as the Landlord under this Lease. Upon such attornment, this Lease shall continue in full force and effect as, or as if it were, a direct lease between the successor landlord and Tenant upon all of the terms, conditions and covenants as are set forth in this Lease and Tenant shall look solely to the successor landlord for the return of the Security Deposit, except that the successor landlord shall not:
(a)be liable for any previous act or omission of Landlord under this Lease, except to the extent that the same is continuing and Tenant has given successor landlord notice thereof;
Page 33 of 60
(b) be subject to any offset not provided for in this Lease, which shall have theretofore accrued to Tenant against Landlord; and
(c) be bound by any previous modification of this Lease not provided for in this Lease, or by any previous prepayment of more than one month’s Base Rent, unless such modification or prepayment shall have been approved in writing by the lessor of such superior lease or the holder of such mortgage through or by reason of which the successor landlord shall have succeeded to the rights of Landlord under this Lease. Landlord shall promptly attempt to obtain the consent of the lessor of each superior lease and the holder of each superior mortgage to any modification of this Lease or prepayment agreed to by Landlord and Tenant if such consent is required under the terms of the superior lease or mortgage, as the case may be.
Section 22.03. Notices to Lessors and Mortgagees. Intentionally omitted
Section 22.04. Subordination. This Lease shall be automatically subordinate to any mortgages that are now, or may hereafter be, placed upon the Premises and to any and all advances to be made thereunder, and to the interest thereon, and to all renewals, replacements and extensions thereof. Any mortgagee may elect to have this Lease constitute a lien prior to its mortgage, and in the event of such election and upon notification by such mortgagee to Tenant to that effect, this Lease shall be deemed prior in lien to such mortgage, whether this Lease is dated prior to or subsequent to the date of such mortgage. Upon the request of Landlord, or any mortgagee, Tenant shall execute whatever instruments may be reasonably required by Landlord or by any mortgagee to carry out the intent of this Section 22.04 and, in addition, shall execute and deliver such further instruments containing modifications of this Lease, so long as such modifications do not increase Tenant’s monetary obligations under this Lease or otherwise materially and adversely affect Tenant’s rights or privileges under this Lease. With respect to Landlord’s mortgagee as of the date hereof, Landlord and Tenant shall execute, concurrent with its execution of this Lease, a subordination non-disturbance and attornment agreement in the form of Exhibit D attached hereto, and Landlord shall provide a Lender-executed copy of the same to Tenant within a reasonable period after the execution of this Lease. Notwithstanding anything to the contrary contained herein, in order for this Lease to be subordinate to any future mortgages, Landlord shall be required to provide Tenant with a subordination non-disturbance and attornment agreement in a form reasonably acceptable to Tenant.
Section 22.05. Remedies. Failure of Tenant to execute any statements, certificates or instruments necessary or desirable to effectuate the foregoing provisions of this Article 22, within ten (10) days after written request so to do by Landlord, followed by a failure of Tenant to execute the same within five (5) days following a second written request, shall constitute a breach of this Lease.
ARTICLE 23
CURING TENANT’S DEFAULTS
Section 23.01. Landlord’s Right to Cure Tenant’s Defaults. If Tenant shall default in the performance of any of Tenant’s obligations under this Lease after the giving of notice and the expiration of the applicable grace period therefor as provided in this Lease (except in the event of an emergency threatening imminent harm to life or property), Landlord, without thereby waiving such default, may (but shall not be obligated to) perform the same for the account and at the expense of Tenant, if such default remain uncured, or Tenant has not commenced a cure and diligently prosecuted the same, for an additional two (2) business days after Landlord shall have given Tenant notice by either facsimile or mail of its intent to effect such cure.
Section 23.02. Landlord’s Expenses of Cure. Bills for any expenses incurred by Landlord in connection with any performance by it for the account of Tenant, and bills for all costs, expenses and
Page 34 of 60
disbursements of every kind and nature whatsoever, including reasonable attorneys’ fees, involved in collecting or endeavoring to collect the Rental or any part thereof not paid when due or enforcing or endeavoring to enforce any rights against Tenant, under or in connection with this Lease, or pursuant to Laws, including any such cost, reasonable expense and disbursement involved in re-entering the Premises, instituting and prosecuting summary proceedings, as well as bills for any property, material, labor, or services provided, furnished, or rendered, by Landlord or at its instance to Tenant, all at the written request of Tenant (all of which expenses shall constitute items of Additional Rent), may be sent by Landlord to Tenant monthly or immediately at Landlord’s option, and shall be due and payable within thirty (30) days following presentation of such bills and any reasonable backup information to Tenant.
ARTICLE 24
ACCESS BY LANDLORD
Section 24.01. Right of Entry. Landlord and Landlord’s agents shall have the right upon at least twenty-four (24) hours prior written notice to enter the Premises at all times during Tenant’s normal business hours to examine the same and to show them to prospective purchasers or mortgagees provided such entry shall not materially interfere with Tenant’s business. Landlord or Landlord’s agents shall have the further right to enter the Premises upon twenty-four (24) hours prior written notice to make such repairs, alterations, improvements or additions as Landlord may deem necessary or desirable to the Premises and/or Building (including, without limitation, such measures as may be necessary or desirable to cure Tenant’s default, as set forth in Section 23.01) and Landlord shall be allowed to take all material into and upon the Premises that may be required therefor without the same constituting an eviction of Tenant in whole or in part, and the Rental shall not abate while such repairs, alterations, improvements, or additions are being made, by reason of loss or interruption of business of Tenant, or otherwise. Landlord may, at reasonable times upon reasonable written notice during the twelve (12) months prior to the Expiration Date, exhibit the Premises to prospective tenants. Landlord shall use reasonable efforts not to unreasonably interfere with Tenant’s business by virtue of the foregoing, and Tenant shall be entitled to have a representative present for any access by Landlord in exercising its rights under this Section 24.01. Landlord shall be permitted to enter the Premises without notice and at all times in connection with an emergency.
Notwithstanding the foregoing, Tenant, at its own expense may, as hereinafter set forth, designate one or more areas of the Premises to be “Secure Areas” (i.e., portions of the Premises to which Landlord shall not have a right of entry or access for any reason whatsoever, except as otherwise provided below). Tenant may, from time to time, exercise its right to create Secure Areas by delivering to Landlord, for Landlord’s written approval, a plan showing the location of any such Secure Areas. Landlord agrees that it will not unreasonably withhold, condition or delay such consent. If Landlord must gain access to a Secure Areas in a non-emergency situation, Landlord shall contact Tenant, and Landlord and Tenant shall arrange a mutually agreed upon time for Landlord to have such access. Landlord shall be accompanied by an employee of Tenant or a party designated by Tenant (the “Escort”). Tenant shall make an Escort available to Landlord during business hours. At all times, Landlord shall comply with all reasonable security measures of the Tenant pertaining to the Secure Areas. If an emergency representing an imminent risk of injury to persons or material property damage in the Building or the Premises, including, without limitation, a suspected fire or flood, requires Landlord to gain access to the Secure Areas, Landlord may enter the Secure Areas without an Escort. If practicable under the circumstances, Landlord shall immediately notify (which may be oral notification) and request that Tenant make an Escort available to Landlord if time permits, and if Tenant shall not make an Escort available to accompany Landlord, then Tenant hereby authorizes Landlord to enter the Secure Areas forcibly or with a master key, and to enter without an Escort. In any such event, except (subject to Section 14.09 of this Lease) to the extent resulting from Landlord’s negligence or willful misconduct, Landlord shall have no liability whatsoever to Tenant, and Tenant shall pay all reasonable expenses incurred by Landlord in repairing or reconstructing any entrance, corridor, door or other portions of the Premises damaged as a result of a
Page 35 of 60
forcible entry by Landlord. Landlord shall have no obligation to provide either janitorial service or cleaning in the Secure Areas unless Tenant shall make arrangements to have an Escort in the Secure Areas at the time such service or cleaning is provided to the remainder of the Premises.
ARTICLE 25
TENANT’S PROPERTY
Section 25.01. Taxes on Tenant’s Property. Tenant shall be responsible for, and shall pay, prior to delinquency, any and all taxes, assessments, levies, fees and other governmental charges of every kind or nature (collectively “Charges”) levied or assessed by and municipal, county, state, federal or other taxing or assessing authority upon, against or with respect to (i) the Premises or any leasehold interest therein, or any use thereof, including, without limitation, any use and/or occupancy tax, (ii) all fixtures, furnishings, equipment, merchandise and personal property of any kind owned by Tenant and placed, installed or located in, within, upon or about the Premises, and (iii) all or any portion of the Rentals payable by Tenant to Landlord; irrespective of whether any of such items described in clauses (i) through (iv) above are assessed as Tenant’s real or personal property, and irrespective of whether any of such items are assessed to or against Landlord or Tenant. If at any time during the Term any of such Charges are not levied and assessed separately and directly to Tenant (for example, if the same are levied or assessed to Landlord, or upon or against the Building and/or the land underlying the Building), Tenant shall pay to Landlord Tenant’s reasonable share thereof as reasonably determined by Landlord.
ARTICLE 26
HOLDING OVER
Section 26.01. Holding Over. (a) If this Lease is not renewed or extended or a new lease is not entered into between the parties, and if Tenant shall then hold over after the Expiration Date or earlier termination of the Lease, irrespective of whether or not Landlord accepts Rental from Tenant for a period beyond the Expiration Date, Tenant’s occupancy of the Premises after the Expiration Date shall be upon all the terms set forth in this Lease except Tenant shall pay on the first day of each month of the holdover period as Base Rent an amount equal to One Hundred Fifty percent (150%) of the monthly installment of the total Rental (i.e., Base Rent and Additional Rent) payable by Tenant during the last year of the Term (i.e., the year immediately prior to the holdover period).
(b) If Tenant shall holdover or remain in possession of any portion of the Premises beyond Thirty (30) days following the Expiration Date, whether or not Landlord accepts any Rental for a period beyond the Expiration Date, Tenant shall be subject not only to summary proceeding and all damages related thereto, but also to any damages arising out of any lost opportunities (and/or new leases) by Landlord to relet the Premises (or any part thereof). All damages to Landlord by reason of such holding over by Tenant may be the subject of a separate action and need not be asserted by Landlord in any summary proceedings against Tenant.
ARTICLE 27
SECURITY
Section 27.01. Security. The Security Deposit is payable by Tenant to Landlord upon execution of this Lease and shall be held by Landlord, together with any security paid to Landlord pursuant to the provisions of Section 18.02 (collectively, the “Security”), as security for the faithful performance of all of the terms, covenants and conditions of this Lease. Landlord shall in no event be obligated to apply the Security to Rental in arrears or damages for Tenant’s default, although Landlord may so apply the Security, at its option. The Security, if not applied toward the payment of Rental in arrears or toward the payment of damages suffered by Landlord by reason of Tenant’s default, shall be returned to Tenant without interest within thirty (30) days following the expiration of this Lease provided that Tenant has
Page 36 of 60
vacated the Premises and delivered possession thereof to Landlord in accordance with the terms of this Lease. Landlord shall not be obligated to keep the Security as a separate fund and may commingle the Security with its own funds. If Landlord applies the Security in whole or in part, Tenant shall, within Ten (10) days of Landlord’s written demand, deposit sufficient funds to maintain the Security Deposit in an amount equal to at least Two (2) month’s Base Rent failure of which shall be deemed a default under this Lease.
ARTICLE 28
LANDLORD’S LIABILITY
Section 28.01. Transfer of Landlord’s Interest. In the event of any transfer or transfers of Landlord’s interest in the Premises, the transferor shall be automatically relieved of any and all obligations on the part of Landlord accruing from and after the date of such transfer, provided that (a) the interest of the transferor, as Landlord, in any funds then in the hands of Landlord in which Tenant has an interest shall be turned over, subject to such interest, to the then transferee; and (b) notice of such sale or other transfer shall be delivered to Tenant. No lessor under a superior lease, holder of a mortgage or beneficiary of a deed of trust to which this Lease is or may be subordinate, shall be responsible for any security received hereunder, unless such lessor, mortgagee or beneficiary of such deed of trust shall have actually received the security.
Section 28.02. Limitation of Liability. Notwithstanding anything to the contrary provided in this Lease, there shall be absolutely no personal liability on the part of Landlord or any officer, director, shareholder, partner, member, employee or agent of Landlord, whether disclosed or undisclosed (or any successor corporate landlord or any partner of any limited or general partnership which may become Landlord or any individual or other entity), with respect to any of the terms, covenants and conditions of this Lease, and Tenant shall look solely to the interest, income or equity of Landlord in the Building for the satisfaction of each and every remedy of Tenant in the event of a breach or default by Landlord of any of the terms, covenants and conditions of this Lease, such exculpation of personal liability to be absolute and without any exception whatsoever. No other property or assets of Landlord, any successor to Landlord, or any officer, director, shareholder, partner, member, employee or agent of Landlord or any successor to Landlord, shall be subject to judgment, levy, execution or other enforcement procedure for the satisfaction of Tenant’s remedies under or with respect to this Lease or the use or occupancy of the Premises. In no event shall Landlord or Tenant be responsible to each other or anyone claiming by, through or under either party for consequential, indirect or punitive damages, except as may be expressly provided for in Section 26.01.
ARTICLE 29
BROKERAGE CLAIMS
Section 29.01. Brokerage Claims. Each party represents and warrants to the other that it has dealt with no broker or agent in connection with this Lease other than Jones Lang LaSalle Brokerage Inc. and Newmark Knight Frank (the “Brokers”) and agrees to indemnify, hold harmless and defend the other party from and against any and all Liabilities arising from or relating to a breach of the foregoing representation and warranty. Landlord shall pay any fee or commission due the Brokers per separate agreement.
ARTICLE 30
PARKING
Section 30.01 Parking. Landlord shall maintain a parking ratio in the parking area of the building at a ratio of four (4) parking space per one thousand (1,000) rentable square feet. All parking spaces shall be for the use of Tenant’s employees and customers and other invitees at no additional charge
Page 37 of 60
to Tenant. All of the foregoing parking spaces shall be on a non-exclusive, “first-come-first-served” basis, however, Nineteen (19) of such parking space shall be reserved for Tenant’s exclusive use, in a location as determined mutually by Landlord and Tenant and as identified on Exhibit E (“Tenant’s Designated Parking”), such location to be relocated if necessary, to a location as determined mutually by Landlord and Tenant. The parking area, driveways, walkways and any other common areas shall be unattended and subject to reasonable Rules and Regulations to be promulgated by Landlord from time to time; provided, however, in the event of a conflict between such Rules and Regulations and this Lease, the terms and conditions of this Lease shall control. Except to the extent caused by the negligence or willful misconduct of the Landlord or its employees, agents or contractors, the use of the parking area shall be at the risk solely of the individual vehicle owners and users, and Landlord shall not be liable for death or injury to persons in connection with any use of the parking area, nor for any loss or damage, by theft, collision, casualty or otherwise, to any vehicle or its contents. Subject to Tenant’s rights as to Tenant’s Designated Parking, on reasonable prior notice to Tenant, Landlord shall have the right to require Tenant’s employees to park in an employee parking area designated by Landlord.
ARTICLE 31
TERMINATION RIGHT
Section 31.01. Termination Right. Notwithstanding anything to the contrary herein, provided and upon condition that Tenant is not in default of this Lease beyond applicable cure periods at the time Tenant delivers the Early Termination Notice or on the Early Termination Date, Tenant shall have a one-time option to terminate this Lease, effective the first day of the ninety-seventh (97th) month after the Commencement Date of this Lease, (such date, the “Early Termination Date”) only exercisable by Tenant giving Landlord written notice of Tenants election to terminate this Lease (the “Early Termination Notice”), which Termination Notice must be received by Landlord on or before the first day of the eighty-fifth (85th) month after the Commencement Date of this Lease, time being of the essence of such date. As consideration for and a condition precedent to Landlord granting to Tenant the option to terminate the Lease as set forth herein, the Termination Notice shall be accompanied by a certified or cashier’s check made payable to the order of the Landlord in the amount of the Termination Payment (as defined below). Failure of Tenant to timely deliver the Early Termination Notice and/or the Termination Payment shall terminate any option or right Tenant may have hereunder. In the event Tenant delivers the Termination Notice and the Termination Payment to Landlord in a timely manner, Tenant shall nonetheless be responsible to continue to comply with all of the terms and conditions and perform all of its obligations contained in the Lease, including, but not limited to, the payment of all Base Rent and Additional Rent and other charges accruing under the Lease through and including the Termination Date. For the purposes hereof, the Termination Payment shall mean an amount equal to the sum of the following: (i) the sum of Three Hundred Twenty-One Thousand Seven Hundred Ninety-Eight and 26/100 Dollars ($321,798.26); plus (ii) the unamortized portion of all of Landlord’s leasing costs, including any costs associated with Landlord’s Work as set forth in Article 6, any allowances, abatements and brokerage commissions in connection with the Lease, and any actual third party out of pocket legal fees and costs incurred with the drafting, negotiation, and completion of this Lease, amortized over the Term at an interest rate of eight percent (8%) (the “Termination Payment”).
ARTICLE 32
MISCELLANEOUS
Section 32.01. Successors and Assigns. All rights and liabilities given to or imposed under this Lease upon the respective parties hereto shall extend to and bind the several respective heirs, executors, administrators, successors, and permitted assigns of such parties, and if there shall be more than one Tenant, they shall all be bound jointly and severally by the terms, covenants and conditions of this Lease.
Page 38 of 60
Section 32.02. Rules and Regulations. Tenant shall comply with and observe all reasonable rules and regulations established by Landlord from time to time and of which Landlord has given Tenant prior written notice. Tenant’s failure to keep and observe such rules and regulations shall constitute a breach of the terms of this Lease in the same manner as if such rules and regulations were contained herein as covenants. In the case of any conflict between the terms and conditions of such rules and regulations and the terms and conditions of this Lease, the terms and conditions of this Lease shall be controlling.
Section 32.03. Joint and Several Liability. If more than one person or entity is executing this Lease as Tenant, each such person or entity shall be jointly and severally liable for the obligations of Tenant under this Lease.
Section 32.04. Tenant Damages. In any instance where the obtaining of Landlord’s consent or approval shall be required under this Lease, the failure of Landlord to give such consent or approval shall not render Landlord liable for damages, and Tenant’s sole remedy in such event shall be an action for specific performance or injunction, and such remedy shall be available only in those cases where Landlord has agreed not to unreasonably withhold its consent or where, as a matter of law Landlord may not unreasonably withhold its consent.
Section 32.05. Covenant of Quiet Enjoyment. Upon payment by Tenant of the Rental and upon the observance and performance of all of the covenants, terms and conditions on Tenant’s part to be observed and performed under this Lease, Tenant shall peaceably and quietly hold and enjoy the Premises for the Term, without hindrance or interruption by Landlord or any other person or persons lawfully or equitably claiming by, through or under Landlord, subject, nevertheless, to the terms and conditions of this Lease, and any mortgage, deed of trust or lease to which this Lease is subordinate.
Section 32.06. Waiver. The subsequent acceptance of Rental by Landlord shall not be deemed to be a waiver of any preceding breach by Tenant of any of the terms, covenants or conditions of this Lease, other than the failure of Tenant to pay the particular Rental so accepted, regardless of Landlord’s knowledge of such preceding breach at the time of acceptance of such Rental. In particular, but without limitation, if Tenant assigns or transfers its interest in this Lease contrary to the terms of this Lease, any acceptance by Landlord of such assignee’s or transferee’s payment shall not be deemed to be a waiver of the restrictions set forth in Articles 17 or 18. One or more waivers of a breach of any covenant or condition shall not be construed as a waiver of a subsequent breach of the same covenant or condition, and the consent to or approval of any act requiring consent or approval shall not be deemed to render unnecessary the obtaining of consent to or approval of any subsequent similar act. The failure of Landlord to insist upon the strict performance of any of the terms, covenants or conditions contained in this Lease shall not be deemed a waiver of any rights or remedies that Landlord may have and shall not be deemed a waiver of any subsequent breach or default in the performance of the terms, covenants or conditions herein contained. No breach by Tenant of a covenant or condition of this Lease shall be deemed to have been waived by Landlord unless such waiver is in writing and signed by Landlord. No act or thing done by Landlord or Landlord’s agents shall be deemed an acceptance of surrender of the Premises and no agreement to accept such surrender shall be valid unless in a writing signed by Landlord.
Section 32.07. Interpretations. Nothing contained herein shall be deemed or construed by the parties hereto, nor by any third party, as creating the relationship of principal and agent or of partnership or of joint venture between the parties hereto, and neither the method of computation of Rental, nor any other provision contained herein, nor any acts of the parties hereto shall be deemed to create any relationship other than the relationship of Landlord and Tenant. Whenever herein the singular number is used, the same shall include the plural, and the masculine gender shall include the feminine and neuter genders.
Page 39 of 60
Section 32.08. Force Majeure. If either party hereto shall be delayed in the performance of its construction or maintenance and/or repair obligations, by reason of strikes, lockouts, labor troubles or inability to procure materials, or shall at any time be so delayed by reason of failure of power, restrictive Laws or reasons of a similar nature not the fault of the party delayed in performing or doing the acts required under this Lease, then performance of such act shall be excused for the period of the delay and the period for the performance of any such act shall be extended for a period equivalent to the period of such delay. In no event shall the provisions of this Section 32.08 operate to excuse Tenant from payment of any Rental.
Section 32.09. Notices. Unless specifically stated to the contrary in this Lease, any notice, demand, request or other instrument which may be or is required to be given by Tenant or Landlord under this Lease or by Laws (“Notices”) shall be in writing and sent by national overnight delivery service or by United States certified mail, return receipt requested, postage prepaid, and shall be deemed to have been given, if sent by national overnight delivery service, as of the first (1st) weekday upon which delivery is first attempted and, if sent by United States Certified Mail, as of the third (3rd) business day following deposit in the United States mail. Notices given in accordance with this Section 32.09 shall be addressed (a) if to Landlord, at the address first set forth hereinabove or at such other address as Landlord may designate by notice given in accordance with this Section 32.09 (in each case with copies delivered by e-mail to: bfishbane@broadmgmt.com), and (b) if to Tenant, at the address set forth for Tenant in the first paragraph of this Lease until Tenant shall take occupancy of the Premises, and thereafter at the Premises, Attn: Michael Kalb and Donna Pasek (in each case with copies delivered by e-mail to: michael.kalb@amarincorp.com and donna.pasek@amarincorp.com) or such other address as Tenant shall designate by notice given in accordance with this Section 32.09. Notices may be given by the attorneys for the respective parties.
Section 32.10. Captions and Section Numbers. The captions, section numbers, article numbers, and index appearing in this Lease are inserted only as a matter of convenience and in no way define, limit, construe, or describe the scope or intent of such sections or articles of this Lease, nor in any way affect this Lease.
Section 32.11. Recording. Tenant shall not record this Lease.
Section 32.12. Furnishing of Financial Statements. Upon Landlord's request (but not more frequently than annually), Tenant shall promptly furnish Landlord with a balance sheet reflecting Tenant's current financial condition, in a form and detail consistent with what was provided to Landlord by Tenant prior to the execution of the Lease.
Section 32.13. Accord and Satisfaction. Payment by Tenant or receipt by Landlord of a lesser amount than the Rental may, at Landlord’s sole option, be deemed to be on account of the earliest due Rental or deemed to be on account of Rental owing for the current period only, notwithstanding any instructions by or on behalf of Tenant to the contrary, which instructions shall be null and void, and no endorsement or statement on any check or any letter accompanying any check or payment as Rental shall be deemed an accord and satisfaction except with Landlord’s written consent, and Landlord shall accept such check or payment without prejudice to Landlord’s right to recover the balance of such Rental or pursue any other remedy in this Lease or at law or in equity against Tenant unless otherwise agreed in writing.
Section 32.14. Execution of Lease; No Option. The submission of this Lease to Tenant shall be for examination purposes only, and does not and shall not constitute a reservation of or option for Tenant to lease, or otherwise create any interest of Tenant in the Premises or any other premises situated in the Building. The return to Landlord of Tenant-executed copies of this Lease shall not be binding upon Landlord, notwithstanding any preparation or anticipatory reliance or expenditures by Tenant or any time
Page 40 of 60
interval, until Landlord has in fact executed and actually delivered a fully-executed copy of this Lease to Tenant.
Section 32.15. Governing Law and Jurisdiction. This Lease shall be governed by and construed in accordance with the laws of the State of New Jersey. If any provision of this Lease or the application thereof to any person or circumstances shall, to any extent, be invalid or unenforceable, the remainder of this Lease shall not be affected thereby and each remaining provision of the Lease shall be valid and enforceable to the fullest extent permitted by Law. Landlord and Tenant hereby irrevocably submit themselves to the exclusive jurisdiction of the state courts of the State of New Jersey and the United States District Court, District of New Jersey in the event of any action or controversy concerning this Lease or the Premises, Building or Property.
Section 32.16. Certain Rules of Construction. Time is of the essence in this Lease. Landlord and Tenant have had substantial experience with the subject matter of this Lease and have each fully participated in the negotiation and drafting of this Lease. Accordingly, this Lease shall be construed without regard to the rule that ambiguities in a document are to be construed against the drafter.
Section 32.17. Authority. The person executing this Lease on behalf of Tenant does hereby represent and warrant to Landlord that Tenant is a duly organized and validly existing corporation, that Tenant has organized in or is qualified to do business in the State of New Jersey, that all Tenant’s franchise taxes have been paid to date, that Tenant has the full right, power and authority to enter into and perform this Lease, and that each person signing this Lease on behalf of Tenant is authorized to do so and to bind Tenant to the terms of this Lease. The person executing this Lease on behalf of Landlord does hereby represent and warrant to Tenant that Landlord is a duly organized and validly existing limited liability company, that Landlord has organized in or is qualified to do business in the State of New Jersey, that Landlord has the full right, power and authority to enter into and perform this Lease, and that each person signing this Lease on behalf of Landlord is authorized to do so and to bind Landlord to the terms of this Lease.
Section 32.18.OFAC. Tenant represents and warrants to Landlord that Tenant is not and shall not become a person or entity with whom Landlord is restricted from doing business under any regulations of the Office of Foreign Asset Control (“OFAC”) of the Department of the Treasury (including, but not limited to, those named on OFAC's Specially Designated and Blocked Persons list) or under any statute, executive order (including, but not limited to, the September 24, 2001, Executive Order Blocking Property and Prohibiting Transactions With Persons Who Commit, Threaten to Commit, or Support Terrorism), or other governmental action and is not and shall not engage in any dealings or transaction or be otherwise associated with such persons or entities.
Section 32.19.Back-Up Generator. Tenant shall have the right, upon Tenants request, to use up to Tenant’s Proportionate Share of capacity of the emergency back-up generator (the “Back-Up Generator”) currently located in the Building. If Tenant so requests, then Tenant may, at its sole cost and expense, tie-into the Back-Up Generator, subject to the reasonable rules and guidelines adopted from time to time by Landlord with respect thereto, and to all applicable laws, codes, regulations and guidelines. Any and all work and improvements to be performed by Tenant to effectuate Tenants tie-in to the Back-Up Generator (such as installing conduits, and connections from the Back-Up Generator to the Premises) shall be considered to be an Alteration, shall be performed in accordance with the provisions of Article 12 of this Lease, and shall be subject to Landlords review and prior written consent in all respects, such approval not to be unreasonably withheld, conditioned or delayed. In the event Tenant elects to tie-into the Back-Up Generator, Tenant shall pay, as Additional Rent, within thirty (30) days after receipt of invoices therefor from Landlord, a pro rata share of the annual fuel and maintenance charges for the Back-Up Generator, which pro rata share shall be based on a ratio, the numerator of which is Tenants total usage of Back-Up Generator capacity and the denominator of which is the aggregate usage of Back-
Page 41 of 60
Up Generator capacity at the applicable period of time. In addition, Tenant, at Tenant’s sole cost and expense, shall be entitled to install its own generator, at a location to be approved by Landlord, such approval not to be unreasonably withheld, conditioned or delayed. Tenant’s installation of such generator shall be in accordance with Article 12 of this Lease and shall be in compliance with all applicable Laws.
Section 32.20.Signatures and Counterparts. This Lease may be executed in two or more counterparts, each of which shall be deemed an original, but all of which together constitute one and the same instrument. The parties may execute and deliver the counterparts of this Lease and any ancillary documents electronically by facsimile or email. The electronic copies of the signatures of the parties will be as valid and binding on the parties as original ink signatures. The receiving party may rely on the receipt of the document executed and delivered electronically as if the original had been received. The parties authorize each other to detach and combine signature pages and consolidate them into a single document. Any one of the completely executed counterparts shall be sufficient proof of this Lease.
ARTICLE 33
Guaranty
Section 33.01. Guaranty. Concurrent with Tenant’s execution and delivery of this Lease and as a material inducement to Landlord’s execution of this Lease, Tenant shall deliver to Landlord a guaranty from Amarin Corporation plc, a company organized under the laws of England and Wales (the “Guarantor”) of Tenant's obligations under this Lease in the form of Exhibit F attached hereto (the “Guaranty”).
[SIGNATURE PAGE TO FOLLOW]
Page 42 of 60
IN WITNESS WHEREOF, Landlord and Tenant have duly executed this Lease as of the date first above written.
|
|
|
Landlord: |
|
|
440 Route 22 LLC |
|
By: /s/David Elkouby |
|
Print Name: David Elkouby |
|
Title: Member |
|
|
|
Tenant: |
|
AMARIN PHARMA, INC. |
|
By: /s/ John Thero |
|
Print Name: John Thero |
|
Title: President and Chief Executive Officer |
|
|
Exhibits to Be Attached:
APremises Cross-Hatched
BPhase One Space Plan
B-1 Building Standards
CPhase Two Space Plan
DSubordination, Non-Disturbance & Attornment Agreement
ETenant’s Designated Parking Plan
FGuaranty Agreement
Schedule 2.05 – Existing Tenants with ROFO Rights
Page 43 of 60
EXHIBIT A
PREMISES CROSS-HATCHED
[Intentionally Omitted]
Page 44 of 60
PHASE ONE FLOOR PLANS
[Intentionally Omitted]
Page 45 of 60
[Intentionally Omitted]
Page 46 of 60
PHASE TWO FLOOR PLANS
[Intentionally Omitted]
Page 47 of 60
SUBORDINATION, NON-DISTURBANCE & ATTORNMENT AGREEMENT
THIS AGREEMENT is made this day of, 2019 by and among
parties referenced below as the Tenant, Lender, and Landlord concerning the Lease, Premises, Mortgage and Property referenced below.
Preliminary Statement
A.The following terms referenced in the introductory paragraph of this document shall have the meanings hereinafter noted:
LANDLORD: 440 Route 22 LLC, a New Jersey limited liability company
TENANT: Amarin Pharma, Inc., a Delaware corporation
LENDER: _____________________, a Delaware limited liability company, its
subsidiaries and/or affiliates, and their respective successors and assigns.
LEASE:Lease dated, ___2019, as nowamended (if
at all), by and between Landlord and Tenant for the Premises.
|
|
PREMISES: |
Suite # ______; approximately67,747 rentable square feet of floor space located at the Property |
MORTGAGE: The Mortgage, Security Agreement and Fixture Filing from
Landlord to Lender, dated and recorded approximately concurrently herewith or, if recording information has been inserted in this
sentence, which Mortgage is dated , 201_ and
recorded with theCounty Registry of Deeds
in Book _____, Page ____.
PROPERTY: The land generally described in Exhibit A attached hereto and the
buildings and improvements thereon now known as
, 440 US
Highway 22, Bridgewater, New Jersey.
B.Landlord and Tenant have entered into the Lease for the Premises located on the Property.
C.Lender is the holder of the Mortgage encumbering the Property and securing a loan from Lender to Landlord (the “Loan”).
D.The parties desire to establish certain rights of quiet and peaceful possession for the benefit of Tenant under the Lease and to define the terms, covenants and conditions precedent for such additional rights;
Page 48 of 60
NOW, THEREFORE, in consideration of the foregoing, the various agreements herein and for other good and valuable consideration, the receipt and sufficiency of which is acknowledged, Landlord, Tenant and Lender covenant and agree as follows:
1.Definitions
As used herein, the terms “Landlord”, “Tenant”, “Lender”, “Lease”, “Premises”, “Mortgage” and “Property”, shall have the meanings ascribed to such terms in subsection A of the Preliminary Statement above. As used herein, “Lender” shall also include the successors and assigns of Lender and any person, party or entity which shall become the owner of the Property by reason of a foreclosure of the Loan Documents or the acceptance of a deed or assignment in lieu of foreclosure or otherwise; “Mortgage” shall also include any renewal, modification, consolidation or extension of the same; “Successor Landlord” shall mean the successors-in-interest to Landlord’s interest under the Lease, including, without limitation, any Lender which shall become the owner of the Property by whatever means; “Property” shall also include improvements hereafter located thereon and the estates therein encumbered by the Loan Documents; “Loan Documents” shall include the Mortgage and all other now or hereafter existing instruments evidencing or securing the Loan; and “Default” shall mean an event of default or an event which, if uncured following the giving of notice or passage of time, or both, would constitute an event of default.
2.Subordination
Subject to the terms of the following Section 3 and other provisions of this Agreement, the Lease, Tenant’s leasehold interest and all rights of Tenant created thereby, and any renewals, extensions, amendments, consolidations, replacements or modifications thereof, to the full extent of all sums due to Lender from Landlord, shall be, and are, completely and unconditionally subject, subordinate and at all times inferior to the lien and provisions of the Loan Documents, and to all of the terms, conditions, and provisions thereof, and all advancements made or to be made thereunder, as same may hereafter be amended, increased, renewed, modified, consolidated, replaced, combined, substituted, severed, split, refinanced, recast, or extended. The Loan Documents shall take priority over the Lease and shall be entitled to the same rights and privileges, both at law and in equity, as the Loan Documents would have had if they had been executed, delivered, and recorded prior to the execution, delivery, or recording of the Lease.
3.Non-Disturbance
As long as the Lease is in full force and effect, and the Tenant is not in default (following any required notices and beyond any period given Tenant in the Lease to cure such default) in the payment of rent, additional rent, or in the performance of any of the terms, covenants, or conditions of the Lease on Tenant’s part to be performed:
a)Tenant’s possession, use and quiet enjoyment of the Property under the Lease as well as Tenant’s
rights and privileges under the Lease, or any extensions or renewals thereof which may be effected in accordance with any renewal rights therefore in the Lease, shall not be diminished or interfered with by Lender;
|
b) |
Tenant’s occupancy of the Property shall not be disturbed by Lender for any reason whatsoever, including any foreclosure proceeding or other action brought pursuant to the Loan Documents; |
|
c) |
the Lease shall not be terminated (except upon its terms or expiration of the term thereof) by Lender; |
Page 49 of 60
4.Tenant Not to be Joined in Foreclosure (Unless Necessary)
As long as Tenant is not in default (following any required notices and beyond any period given Tenant in the Lease to cure such default) in the payment of rents, additional rent, or in the performance of any of the other terms, covenants, or conditions of the Lease on Tenant’s part to be performed, Lender will not join Tenant as a defendant in any action or proceeding foreclosing the Loan Documents unless (i) such joinder is necessary to foreclose the Loan Documents, and then only for such purpose and not for the purpose of terminating the Lease, or (ii) Tenant is the Borrower or a guarantor of the Loan, in whole or part.
5.Tenant to Attorn to Successor Landlord
If the interest of Landlord shall be transferred to and owned by Lender or by any and all successors-in-interest to Lender or such other purchaser by reason of foreclosure, accepting a deed in lieu of foreclosure, or other proceedings, or some similar doctrine, brought by it in lieu of Landlord under the Lease, Tenant shall be bound to Successor Landlord under all of the terms, covenants, and conditions of the Lease for the balance of the term thereof remaining, and any extensions or renewals thereof which may be effected in accordance with any option therefore in the Lease, with the same force and effect as if Successor Landlord were the Landlord under the Lease, and Tenant does hereby, and will, affirm its obligations under the Lease, attorn to Successor Landlord as its landlord, without the execution of any further instruments, provided, Tenant shall be under no obligation to pay rent to Successor Landlord until Tenant receives written notice from Successor Landlord that Landlord is in default under the Loan Documents and/or that it has succeeded to the interests of Landlord under the Lease. The consent and approval of Landlord to this Agreement shall constitute an express authorization for Tenant to make such payment to Successor Landlord and Landlord waives any claim against Tenant for any rent payments made to Successor Landlord at Successor Landlord’s direction. The respective rights and obligations of Tenant and Successor Landlord upon such attornment, to the extent of the then remaining balance of the term of the Lease and any such extensions and renewals, shall be and are the same as now set forth therein, subject to paragraph 6 below, as well as paragraph 9 below concerning purchase options; it being the intention of the parties hereto for this purpose to incorporate the Lease in this Agreement by reference with the same force and effect as if set forth herein, subject to paragraph 6 below, as well as paragraph 9 below concerning purchase options.
Tenant agrees to execute and deliver at any time and from time to time upon the request of Landlord or any Successor Landlord (i) any reasonable instrument or certificate, as the case may be, reasonably deemed to be necessary or appropriate to evidence such attornment; and (ii) a replacement lease for the balance of the term of the Lease on the same terms and conditions as the Lease. Tenant waives the provisions of any statute or rule of law now or hereafter in effect that may give or purport to give it any right or election to terminate or otherwise adversely affect the Lease or the obligations of Tenant thereunder by reason of any foreclosure of similar proceeding.
6.Successor Landlord Not Bound by Certain Actions of Landlord
In the event that any Successor Landlord succeeds to the interest of Landlord under the Lease, such Successor Landlord shall not be:
a) liable for any act or omission of any prior landlord (including Landlord), or to cure any default of any prior landlord (including Landlord) except to the extent that the same is continuing and Tenant has given Successor Landlord notice thereof;
b) subject to any offsets, defenses or claims which Tenant may have against any prior landlord (including Landlord); except for offset rights available under the Lease;
Page 50 of 60
c) bound by any rent or additional rent which Tenant may have paid for more than one (1) month in advance, or for any security or others deposit, whether or not still held by any prior landlord (including Landlord), unless such rent, additional rent or any security or other deposit was actually received by such Successor Landlord;
d) bound by any agreement, amendment, modification, cancellation or termination of the Lease which was made without the prior written consent of Successor Landlord which consent may be withheld, conditioned, or delayed for any reason, in the sole discretion of Successor Landlord;
e) obligated to complete any construction work required to be done by Landlord pursuant to the
provisions of the Lease or to reimburse Tenant for any construction work done by Tenant, Tenant hereby acknowledging that Landlord has no obligation to reimburse Tenant for any work done by Tenant;
f) required to make any general repairs to the Property, or to the Property required as a result of
fire or other casualty, or by reason of condemnation, unless Successor Landlord shall be obligated under the Lease to make such repairs and Successor Landlord shall have received sufficient casualty insurance proceeds or condemnation awards to finance the completion of such repairs;
g) required to make any capital improvements to the Property in general or to the Property which Landlord may have agreed to make, but had not completed;
h) liable or responsible for payment of any brokerage or other commission or compensation due with respect to the Lease or any renewal, extension, expansion or other amendment thereof;
i) liable to Tenant for any actions of its successors-in-interest upon Successor Landlord’s
subsequent transfer of its interest in the Property.
Notwithstanding anything to the contrary, nothing contained herein shall limit Tenant’s rights, defenses or remedies against a prior landlord (including Landlord) for any default by a prior landlord (including Landlord) under the Lease which remains uncured following the time Lender or any Successor Landlord succeeds in interest to Landlord under the Lease, nor relieve Lender or any Successor Landlord of the obligation to cure ongoing defaults that are continuing following the date that Lender or any Successor Landlord succeeds in the interest of Landlord under the Lease, provided that Lender or Successor Landlord (as applicable) is given written notice of such default and thereafter fails to cure the same within the time period set forth in this Agreement.
7. Notices
a)Tenant shall promptly deliver written notice to Lender of any default of Landlord which would entitle Tenant to cancel the Lease or abate the rent payable thereunder. Lender shall have the right to cure such default within thirty (30) days. Tenant further agrees not to invoke any of its remedies
under the Lease against Lender until the requisite time period has elapsed, or if the default cannot be reasonably cured within such time period, such longer period of time as may be reasonably necessary to cure such default, so long as Lender commences efforts to cure such default and prosecute such efforts with reasonable diligence including, without limitation, such time as may be necessary to foreclose on its Loan Documents, judicially, or by power of sale.
b)Any notice, direction, demand, request, permission, approval, consent, election or other communications given or made under this Agreement shall be in writing and shall be hand delivered or sent by FedEx or other reputable nationally recognized overnight courier service, and shall be deemed given when received at the following addresses:
Page 51 of 60
c/o Shem Creek Capital
16 Laurel Avenue, Suite 20
Wellesley, Massachusetts 02481
Attn: Scott GoldbergAttn:
With copy to:If to Tenant:
Bernkopf Goodman, LLP
Two Seaport Lane
Boston, Massachusetts 02210
Attn: David L. Doyle, Esquire
8.Lease Termination
In the event Tenant exercises any right it may have to terminate the Lease by paying a lease cancellation fee, termination fee, surrender fee, settlement amount, accelerated rent or other such payment, as specified under the terms of the Lease (hereafter, the “Lease Termination Fee”), Tenant agrees to deliver such Lease Termination Fee to Lender or Successor Landlord. Lender or Successor Landlord shall hold such Lease Termination Fee in accordance with the terms of the Loan Documents. Any lease termination notice which must be given by Tenant to Landlord shall be given to Landlord or Successor Landlord, whichever is applicable, and to Lender.
9.Purchase Options
Any options or rights contained in the Lease to acquire title to the Property are hereby made subject and subordinate to the rights of Lender or Successor Landlord under the Loan Documents. Any right of Tenant to purchase the Property, including any right of first refusal, right of first offer, or similar provisions, shall not apply to a foreclosure sale of the Property by Lender pursuant to its rights under the Loan Documents and shall be extinguishable by any foreclosure of the Loan Documents. Any right of Tenant to cancel the Lease in order to move to other property to be leased or purchased from Landlord and any right of Tenant to inducements to be provided by Landlord but not set forth in the Lease shall be extinguishable by any foreclosure of the Loan Documents.
10.Assignment of Lease
Notwithstanding that the Lease is being assigned by Landlord to Lender under the Loan Documents (which assignment Landlord hereby acknowledges), Lender assumes no duty, liability or obligation whatsoever under the Lease. All rent payments under the Lease shall continue to be paid to Landlord in accordance with the terms of the Lease unless and until Lender or Successor Landlord directs Tenant otherwise in writing, in which event, Tenant shall pay all future rent as directed. Under the provisions of the assignment, Tenant understands that, without the consent of Lender or Successor Landlord, the Lease cannot be amended in any respect or terminated (either directly or by the exercise of any option which could lead to termination) and consent cannot be given by Tenant to the release of any party having liability thereon.
11.Successors and Assigns
Page 52 of 60
The terms of this Agreement shall be binding upon, inure to the benefit of, and be enforceable by, the parties and their respective heirs, administrators, legal representatives, successors and assigns.
12.Miscellaneous
This Agreement (i) contains the entire agreement with respect to the subject matter hereof; (ii) may not be modified or terminated, nor may any provisions hereof be waived, orally or in any manner other than by an agreement in writing signed by the parties hereto or their respective successors, administrators, or assigns; (iii) shall inure to the benefit of, and be binding upon, the parties hereto, their successors and assigns and any purchaser and its or their respective heirs, personal representatives, successors and assigns (including, without limitation, (a) Tenant’s permitted assignees and (b) any purchaser of the Property at a foreclosure sale or any grantee under a deed in lieu of foreclosure). This Agreement may be executed in any number of counterparts, each of which shall be effective only upon delivery and thereafter shall be deemed an original, and all of which shall be taken to be one and the same instrument, for the same effect as if all parties hereto had signed the same signature page.
13. Applicable Law
This Agreement shall be governed by and construed in accordance with the laws of the state in which the Property is situated in connection with any action, claim or proceedings related to the Property.
EXECUTED as a sealed instrument as of the day and year first above written.
Witnesses:LANDLORD:
440 ROUTE 22 LLC
__________________________ By:
By:______________________________________
Its:_____________________________________
Duly Authorized
Witnesses:TENANT:
AMARIN PHARMA, INC.
____________________________ By:______________________________________
Its:______________________________________
Duly Authorized
Witnesses:LENDER:
_____________________________ By:______________________________________
Its:_____________________________________
Duly Authorized
Page 53 of 60
On this _____ day of, 201_ before me, the undersigned notary public, personally
appeared, proved to me through satisfactory evidence of identification,
which was, to be the person whose name is signed on the preceding or attached document, and acknowledged to me that he/she signed it voluntarily for its stated purpose.
NOTARY PUBLIC
[Affix Notarial Seal]
Printed Name:
My Commission Expires:
STATE OF
, ss.
On this _____ day of, 201_ , before me, the undersigned notary public, personally
appeared, proved to me through satisfactory evidence of identification,
which was, to be the person whose name is signed on the preceding or
attached document, ason behalf of the Landlord, and acknowledged to
me that he/she signed it voluntarily for its stated purpose.
NOTARY PUBLIC
[Affix Notarial Seal]
Printed Name:
My Commission Expires:
STATE OF
, ss.
On this _____ day of, 201__, before me, the undersigned notary public,
personally appeared, proved to me through satisfactory evidence of
identification, which was, to be the person whose name is signed on the
preceding or attached document, as line on behalf of the Lender, and acknowledged to me that he/she signed it voluntarily for its stated purpose.
NOTARY PUBLIC
[Affix Notarial Seal]
Printed Name:
My Commission Expires:
Page 54 of 60
[Legal Description of Property Attached Hereto]
Page 55 of 60
EXHIBIT E
TENANT’S DESIGNATED PARKING PLAN
[Intentionally Omitted]
Page 56 of 60
GUARANTY
Amarin Corporation plc, a company organized under the laws of England and Wales, with an office at 2 Pembroke House, Upper Pembroke Street 28-32, Dublin 2, Ireland (“Guarantor”) has requested that 440 Route 22 LLC (“Landlord”) to enter into a Lease of even date herewith (the “Lease”) with Amarin Pharma, Inc., a Delaware corporation (“Tenant”), covering certain premises located at 440 US Highway 22, Bridgewater, New Jersey, as more particularly described in the Lease. In order to induce Landlord to enter into the Lease and in consideration of Landlord's entering into the Lease, Guarantor hereby guarantees, unconditionally and absolutely, to Landlord, its successors and assigns (without requiring any notice of non-payment, non-keeping, non-performance or non-observance or proof of notice or demand whereby to charge Guarantor all of which Guarantor hereby waives) the full and faithful keeping, performance and observance of all the covenants, agreements, terms, provisions and conditions provided to be kept by Tenant under the Lease, including, without limitation, the payment as and when due of all Base Rent, Additional Rent, charges and damages payable by Tenant under the Lease, and the payment of any and all other damages for which Tenant shall be liable by reason of any act or omission contrary to any of such covenants, agreements, terms, provisions or conditions. All capitalized terms not defined herein shall have the meanings ascribed to them in the Lease.
As a further inducement to Landlord to enter into the Lease and in consideration thereof, Guarantor hereby covenants and acknowledges as follows:
(1)Guarantor is the record and beneficial owner of an equity interest in Tenant.
(2)The obligations of Guarantor shall not be terminated or affected in any way or manner whatsoever by reason of Landlord's failure to resort to any summary or other proceedings, actions or remedies for the enforcement of any of Landlord's rights under the Lease or by reason of any extensions of time or indulgences granted by Landlord, or by reason of the assignment or surrender of all or any part of the Lease or the term and estate thereby granted or all or part of the Premises. The liability of Guarantor is co-extensive with that of Tenant and also joint and several, and action or suit may be brought against Guarantor and carried to final judgment and/or completion and recovery had, either with or without making Tenant or any other guarantor a party thereto. Insofar as the payment by Tenant of any sums of money to Landlord is involved, this Guaranty is a guaranty of payment and not of collection and shall remain in full force and effect until payment in full to Landlord of all sums payable under this Guaranty. Guarantor waives any right to require that any action be brought against Tenant or to require that resort be had to any secured interest, security or to any other credit in favor of Tenant.
(3)If, pursuant to law or to any option granted by the Lease, the Lease shall be renewed or its terms extended for any period beyond the initial termination date of the Lease, or if the Lease be modified by agreement between Landlord and Tenant in any other respect, the obligations of Guarantor shall extend and apply with respect to the full and faithful keeping, performance and observance of all of the covenants, agreements, terms, provisions and conditions which under such renewal of the Lease or extension of its terms or which under any new lease or amendment or modification agreement, entered into for the purpose of express or confirming any such renewal, extension, inclusion, substitution or modification, are to be kept, performed and observed by Tenant (including, without being limited to, the payment as and when due of rent, additional rent, charges and damages provided for thereunder) and the payment of any and all after damages for which Tenant shall be liable by reason of any act or omission contrary to any of such covenants, agreements, terms, provisions or conditions.
Page 57 of 60
(4) Neither Guarantor's obligation to make payment in accordance with the terms of this Guaranty nor any remedy for the enforcement thereof shall be impaired, modified, changed, stayed, released or limited in any manner whatsoever by any impairment, modification, change, release, limitation or stay of the liability of Tenant or its estate in bankruptcy or any remedy for the enforcement thereof, resulting from the operation of any present or future provision of the Bankruptcy Act of the United States or other statute or from the decision of any court interpreting any of the same, and Guarantor shall be obligated under this Guaranty as if no such impairment, stay, modification, change, release or limitation has occurred.
(5)This Guaranty shall be binding on Guarantor and its successors and assigns and inure to the benefit of Landlord and its successors and assigns.
(6)Guarantor waives the right to trial by jury in any action or proceeding in respect of this Guaranty.
(7)It is expressly understood and agreed by Guarantor and Landlord that all matters arising out of the Lease and this Guaranty, including the validity or any provisions hereof, are to be governed by, interpreted and construed in accordance with the laws of the State of New Jersey (without giving regard or effect to any conflicts of law rules or other choice of law rules).
(8)With respect to any dispute or legal action of any kind arising from the terms of this Guaranty that any party may have, either during the term of this Guaranty or thereafter, it is expressly agreed that such action shall be brought either in the state courts of the State of New Jersey (or in the United States District Court for the District of New Jersey, to the extent such court has jurisdiction thereof), and that such court shall be deemed to be the court of sole and exclusive jurisdiction and venue for the bringing of such action. The foregoing consent to jurisdiction and venue shall not constitute general consent by Guarantor to jurisdiction and venue in the State of New Jersey for any purpose except as provided herein and shall not be deemed to confer rights on any other person or entity.
(9)Guarantor consents that Tenant shall hereafter have full authority and be duly empowered to accept service of process on behalf of Guarantor in connection with the enforcement of this Guaranty, and Guarantor hereby appoints Tenant as its agent for purposes of acceptance of service of process in connection with the enforcement of this Guaranty, so long as a copy of any such legal proceeding served upon Guarantor through Tenant is promptly furnished to Guarantor by an international courier service at the following address: 2 Pembroke House, Upper Pembroke Street 28-32, Dublin 2, Ireland.
(10)Guarantor shall pay to Landlord all of Landlord's reasonable expenses, including, without limitation, reasonable third party out of pocket attorneys' fees and disbursements, in enforcing this Lease Guaranty following an event of default by Tenant under the Lease, beyond any applicable notice and cure periods.
[SIGNATURE PAGE TO FOLLOW]
Page 58 of 60
Dated: _______________, 2019
Witness:Amarin Corporation plc,
|
A company organized under the laws of England and Wales |
By: __________________________
|
Name: |
Name: |
Title:
Page 59 of 60
EXISTING TENANTS WITH ROFO RIGHTS
|
|
• |
MOLEX, LLC (assignee) |
|
|
• |
KRAMER LABORATORIES, INC. |
Page 60 of 60
EXHIBIT 21.1
Subsidiaries of the Registrant as of December 31, 2018
|
|
|
|
Name
|
Jurisdiction
|
|
Amarin Pharmaceuticals Ireland Limited |
Ireland |
|
Amarin Pharma, Inc. |
Delaware |
|
Amarin Neuroscience Limited |
Scotland |
|
Corsicanto DAC (liquidated in January 2019) (formerly Corsicanto Limited) |
Ireland |
|
Corsicanto II DAC |
Ireland |
|
Ester Neurosciences Limited |
Israel |
EXHIBIT 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We consent to the incorporation by reference in the following Registration Statements:
|
(1) |
Registration Statement (Form F-1 No. 333-163704) of Amarin Corporation plc, |
|
(2) |
Registration Statement (Form S-8 Nos. 333-146839, 333-143358, 333-132520, 333-110704, 333-101775, and 333-168055) pertaining to the 2002 Stock Option Plan of Amarin Corporation plc, |
|
(3) |
Registration Statement (Form S-8 No. 333-168054) pertaining to the 2008 Long Term Incentive Award dated May 20, 2008 issued to Mr. Tom Maher, Mr. Alan Cooke, and Dr. Declan Doogan of Amarin Corporation plc |
|
(4) |
Registration Statement (Form S-8 Nos. 333-176877, 333-183160, 333-205863 and 333-219644) pertaining to the 2011 Stock Incentive Plan of Amarin Corporation plc, |
|
(5) |
Registration Statement (Form S-8 No. 333-180180) pertaining to the Employment Inducement Award of Amarin Corporation plc, |
|
(6) |
Registration Statement (Form S-8 No. 333-84152), |
|
(7) |
Registration Statement (Form S-3 No. 333-216384) of Amarin Corporation plc, and |
Registration Statement (Form S-3 No. 333-216385) of Amarin Corporation plc;
(8)Registration Statement (Form S-3 No. 333-216385) of Amarin Corporation plc;
of our reports dated February 27, 2019, with respect to the consolidated financial statements of Amarin Corporation plc, and the effectiveness of internal control over financial reporting of Amarin Corporation plc included in this Annual Report (Form 10-K) of Amarin Corporation plc for the year ended December 31, 2018.
/s/ Ernst & Young LLP
Iselin, New Jersey
February 27, 2019
EXHIBIT 31.1
CERTIFICATION
I, John F. Thero, certify that:
|
1. |
I have reviewed this Annual Report on Form 10-K of Amarin Corporation plc; |
|
2. |
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
|
3. |
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
|
4. |
The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
|
|
a. |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
|
|
b. |
Designed such internal controls over financial reporting, or caused such internal controls over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
|
|
c. |
Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
|
|
d. |
Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
|
5. |
The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
|
|
a. |
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
|
|
b. |
Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
|
|
|
|
Date: February 27, 2019 |
/s/ John F. Thero
|
|
|
John F. Thero |
|
|
President and Chief Executive Officer (Principal Executive Officer) |
EXHIBIT 31.2
CERTIFICATION
I, Michael W. Kalb, certify that:
|
1. |
I have reviewed this Annual Report on Form 10-K of Amarin Corporation plc; |
|
2. |
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
|
3. |
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
|
4. |
The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have: |
|
|
a. |
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
|
|
b. |
Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
|
|
c. |
Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
|
|
d. |
Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
|
5. |
The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
|
|
a. |
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
|
|
b. |
Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
|
|
|
|
Date: February 27, 2019 |
/s/ Michael W. Kalb |
|
|
Michael W. Kalb |
|
|
Senior Vice President and Chief Financial Officer |
|
|
(Principal Financial Officer and Principal Accounting Officer) |
EXHIBIT 32.1
STATEMENT PURSUANT TO 18 U.S.C. § 1350
Pursuant to the requirement set forth in Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350), John F. Thero, President and Chief Executive Officer (Principal Executive Officer) of Amarin Corporation plc (the “Company”), and Michael W. Kalb, Senior Vice President and Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) of the Company, each hereby certifies that, to the best of his knowledge:
|
(1) |
The Company’s Annual Report on Form 10-K for the period ended December 31, 2018, to which this Certification is attached as Exhibit 32.1 (the “Annual Report”) fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and |
|
(2) |
The information contained in the Annual Report fairly presents, in all material respects, the financial condition and results of operations of the Company at the end of such year. |
|
|
|
|
|
/s/ John F. Thero
|
|
Date: February 27, 2019 |
John F. Thero |
|
|
President and Chief Executive Officer (Principal Executive Officer) |
|
|
|
|
|
/s/ Michael W. Kalb
|
|
Date: February 27, 2019 |
Michael W. Kalb |
|
|
Senior Vice President and Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) |
This certification accompanies the Form 10-K to which it relates, is not deemed filed with the Securities and Exchange Commission and is not incorporated by reference into any filing of Amarin Corporation plc under the Securities Exchange Act of 1934, as amended (whether made before or after the date of the Form 10-K), irrespective of any general incorporation language contained in such filing.